Input file
Description of the input file, cryspy.in.
Description of the input file, cryspy.in.
CrySPY uses the configparser module to read input file, cryspy.in .
cryspy.in consists of sections, led by a [section] header and followed by name = value or name : value entries.
Section names and values are case sensitive, but names are not.
Lines beginning with # or ; are ignored and may be used to provide comments.
Accepted bool values are 1, yes, true, and on, which cause this method to return True,
and 0, no, false, and off, which cause it to return False.
These string values for bool are checked in a case-insensitive manner.
Some values are given in a space-separated manner.
See configparser in detail.
section name: case sensitive
name: case insensitive
value: case sensitive except for bool
2025 March 6 updated
| Name | Value | Default | Description |
|---|---|---|---|
algo | RS, EA, EA-vc, BO, LAQA | Algorithm | |
calc_code | VASP, QE, OMX, soiap, LAMMPS, ASE | Caluculation code for structure optimization | |
tot_struc | int | The total number of structures. Not used in the case of EA or EA-vc. | |
nstage | int | The number of stages | |
njob | int | The number of jobs running at the same time. | |
jobcmd | str | Command to submit jobs such as qsub and sbatch. | |
jobfile | str | File name of the job file. |
2025 July 4, updated
| Name | Value | Default | Description |
|---|---|---|---|
struc_mode | crystal, mol, mol_bs | crystal | Structure generation mode |
atype | atomic symbol [atomic symbol …] | Atom type. e.g. atype = Na Cl. | |
nat | int [int …] | The number of atoms corresponding to each atype. e.g. nat = 8 8. Not used in EA-vc. | |
mindist (mindist_?) | float [float …] | None | Constraint on minimum interatomic distance [Å]. |
mindist_factor | float | 1.0 | Scaling factor for mindist. |
vol_factor | float | 1.0 | Volume scaling factor. |
vol_mu | float | None | Mean of volume if you want specify the volume of cells. |
vol_sigma | float | None | Standard deviation of volume if you want specify the volume of cells. |
symprec | float | 0.01 | Precision for symmetry finding. |
spgnum | all, space group number, 0 | all | Constraint on space group. If all, 1–230. If 0, random structure without space group information (no symmetry). |
use_find_wy | bool | False | Structure generation with find_wy. |
if algo is EA-vc
| Name | Value | Default | Description |
|---|---|---|---|
ll_nat | int [int …] | Lower limit of nat. e.g. ll_nat = 0 0. | |
ul_nat | int [int …] | Upper limit of nat. e.g. ul_nat = 8 8. | |
charge | int [int …] | None | Used to impose the charge neutrality condition. Charge of each atom type. e.g. charge = 1 -1 for NaCl. |
if struc_mode is mol or mol_bs
| Name | Value | Default | Description |
|---|---|---|---|
mol_file | str [str …] | Path of molecule files or molecule names. | |
nmol | int [int …] | The number of molecules. | |
timeout_mol | float | None | Time out for molecular structure generation. |
rot_mol | random, random_mol, random_wyckoff | random_wyckoff | Only used in mol_bs. Mode for rotation of molecules. |
nrot | int | 20 | Only used in mol_bs. Maximum number of trials to rotate molecules. |
mindist_mol_bs (mindist_mol_bs_?) | float [float …] | None | Only used in mol_bs. Constraint on minimum intermolecular distance [Å]. |
mindist_mol_bs_factor | float | 1.0 | Only used in mol_bs. Scaling factor for mindist_mol_bs. |
if use_find_wy is True or spgnum = 0
| Name | Value | Default | Description |
|---|---|---|---|
fwpath | str | None | Only used with find_wy. Path of find_wy. If None, fwpath is automatically searched in your $PATH. |
minlen | float | Only used with find_wy or spgnum = 0. Minimum length of lattice vector [Å]. | |
maxlen | float | Only used with find_wy or spgnum = 0. Maximum length of lattice vector [Å]. | |
dangle | float | Only used with find_wy or spgnum = 0. Delta angle for alpha, beta, and gamma in degree unit. | |
maxcnt | int | 50 | Only used with find_wy or spgnum = 0. Maximum number of trials to determine atom positions. |
2025 July 12, updated
[VASP] section is required only if you use VASP (calc_code = VASP)
| Name | Value | Default | Description |
|---|---|---|---|
kppvol | int [int …] | Grid density per Å**(-3) of reciprocal cell in each stage. | |
force_gamma | bool | False | If true, force gamma-centered mesh. |
vasp_MAGMOM | float [float …] | None | Used only for EA-vc. MAGMOM values for each element. |
vasp_LDAUL | int [int …] | None | Used only for EA-vc. LDAUL values for each element. |
vasp_LDAUU | float [float …] | None | Used only for EA-vc. LDAUU values for each element. |
vasp_LDAUJ | float [float …] | None | Used only for EA-vc. LDAUJ values for each element. |
[QE] section is required only if you use QE (calc_code = QE)
| Name | Value | Default | Description |
|---|---|---|---|
kppvol | int [int …] | Grid density per Å**(-3) of reciprocal cell in each stage | |
qe_infile | str | File name of QE input file. | |
qe_outfile | str | File name of QE output file. | |
pv_term | bool | False | If true, read enthalpy instead of total energy. |
[OMX] section is required only if you use OpenMX (calc_code = OMX)
| Name | Value | Default | Description |
|---|---|---|---|
kppvol | int [int …] | Grid density per Å**(-3) of reciprocal cell in each stage | |
OMX_infile | str | File name of OpenMX input file. | |
OMX_outfile | str | File name of OpenMX output file. | |
ValenceElectrons | str float float [str float float …] | The number of initial charges for up and down spin states. |
e.g. in NaCl: ValenceElectrons = Na 4.5 4.5 Cl 3.5 3.5.
[soiap] section is required only if you use soiap (calc_code = soiap)
| Name | Value | Default | Description |
|---|---|---|---|
soiap_infile | str | File name of soiap input file. | |
soiap_outfile | str | File name of soiap output file. | |
soiap_cif | str | File name of soiap CIF-formatted initial structure. |
[LAMMPS] section is required only if you use LAMMPS (calc_code = LAMMPS)
| Name | Value | Default | Description |
|---|---|---|---|
lammps_infile | str | File name of LAMMPS input file. | |
lammps_outfile | str | File name of LAMMPS output file. | |
lammps_potential | str [str …], None | None | Potential. |
lammps_data | str | File name of LAMMPS data file. |
[ASE] section is required only if you use ASE (calc_code = ASE)
| Name | Value | Default | Description |
|---|---|---|---|
ase_python | str | File name of ASE input file. |
2025 July 7, updated
| Name | Value | Default | Description |
|---|---|---|---|
n_pop | int | Population (see also Population size) | |
n_crsov | int | Number of offspring created by crossover | |
n_perm | int | Number of offspring created by permutation | |
n_strain | int | Number of offspring created by strain | |
n_rand | int | Number of structures created randomly | |
n_elite | int | Number of elite individuals (see also Natural selection) | |
fit_reverse | bool | False | If False, minimal search (see also Evaluate fitness) |
n_fittest | int | 0 | Number of individuals that remain natural selection. If set to 0, all individuals are retained. |
slct_func | TNM, RLT | Function to select parents | |
t_size | int | 3 | Tournament size. Used only used slct_func = TNM. (see also Tournament selection) |
a_rlt | float | 10.0 | Parameter for linear scaling. Used only with slct_func = RLT. (see also Roulette selection) |
b_rlt | float | 1.0 | Parameter for linear scaling. Used only with slct_func = RLT. (see also Roulette selection) |
crs_lat | equal, random | random | How to mix lattice vectors (see also crossover > 5. Swap the sliced halves) |
nat_diff_tole | int | 4 | Tolerance for difference in the number of atoms in crossover. (see also crossover > 6. Select the offspring with more atoms) |
ntimes | int | 1 | Number of times in permutation. |
sigma_st | float | 0.5 | Standard deviation for strain. |
maxcnt_ea | int | 50 | Maximum number of trials in EA. |
maxgen_ea | int | 0 | Maximum generation. If set to 0, no upper limit is applied. |
emax_ea | float | None | Energy upper limit (eV/atom) for natural selection. |
emin_ea | float | None | Energy lower limit (eV/atom) for natural selection. |
emax_ea and emin_ea are used not in natural selection but when computing the convex hull.| Name | Value | Default | Description |
|---|---|---|---|
n_add | int | Number of offspring created by addition. | |
add_max | int | 3 | (since version 1.4.1) Maximum number of atoms to add in addition |
n_elim | int | Number of offspring created by elimination. | |
elim_max | int | 3 | (since version 1.4.1) Maximum number of atoms to eliminate in elimination |
n_subs | int | Number of offspring created by substitution. | |
subs_max | int | 3 | (since version 1.4.1) Maximum number of atoms to substitute in substitution |
target | str | random | Target. Only random for now. |
end_point | (float, …, float) | Energy of end points for formation energy. | |
emax_ea | float | None | Energy upper limit (eV/atom) for computing the convex hull. |
emin_ea | float | None | Energy lower limit (eV/atom) for computing the convex hull. |
cgen | int | None | (since version 1.4.1) Which generation’s data to plot up to. If None, data will be plotted up to the latest generation. |
show_max | float | 0.2 | When plotting the convex hull, the maximum value of the y-axis (for binary systems) or the maximum hull distance (for ternary systems) is set by show_max. |
lable_stable | bool | True | Whether to show stable compositions when plotting the convex hull. |
vmax | float | 0.2 | Maximum value of the colorbar representing hull distance. |
bottom_margin | float | 0.02 | Bottom margin of the y-axis for binary convex hull plot. |
fig_format | str | svg | Figure format for convex hull plot: svg, png, or pdf. |
2024 May 27th, updated
[BO] section is required only if you use BO (algo = BO)
| Name | Value | Default | Description |
|---|---|---|---|
nselect_bo | int | The number of structures to be selected at once. | |
score | TS, EI, PI | Acquisition function. | |
num_rand_basis | int | 0 | If 0, Gaussian process. The number of basis function. |
cdev | float | 0.001 | Cutoff of deviation for standardization. |
dscrpt | FP | Structure descriptor. | |
max_select_bo | int | 0 | Maximum number of selection. |
manual_select_bo | int [int …] | None | Structure IDs to be selected manually. |
emax_bo | float | None | Upper limit of energy in BO. |
emin_bo | float | None | Lower limit of energy in BO. |
if decrpt is FP
CrySPY 1.3.0 or later
fppath and fp_rmin are obsolete.
| Name | Value | Default | Description |
|---|---|---|---|
fp_rmax | float | 8.0 | Only used with dscrpt = FP. Maximum cutoff of r in fingerprint. |
fp_npoints | int | 20 | Only used with dscrpt = FP. Number of discretized points for each pair in fingerprint. |
fp_sigma | float | 0.7 | Only used with dscrpt = FP. Sigma parameter [Å] in Gaussian smearing function. |
CrySPY 1.2.5 or earlyer
| Name | Value | Default | Description |
|---|---|---|---|
fppath | str | None | Only used with dscrpt = FP. Path of cal_fingerprint. If None, fwpath is automatically searched in your $PATH. |
fp_rmin | float | 0.5 | Only used with dscrpt = FP. Minimum cutoff of r in fingerprint. |
fp_rmax | float | 5.0 | Only used with dscrpt = FP. Maximum cutoff of r in fingerprint. |
fp_npoints | int | 20 | Only used with dscrpt = FP. Number of discretized points for each pair in fingerprint. |
fp_sigma | float | 1.0 | Only used with dscrpt = FP. Sigma parameter [Å] in Gaussian smearing function. |
[LAQA] section is required only if you use LAQA (algo = LAQA)
| Name | Value | Default | Description |
|---|---|---|---|
nselect_laqa | int | The number of structures to be selected at once. | |
wf | float | 0.1 | Weight of the force term. |
ws | float | 10.0 | Weight of the stress term. |
See also Search algorithms > LAQA
If algo = LAQA, the followings are automatically set in the [option] section.
Force and stress data are collected step by step. Energy and structure data are NOT. They are collected for each selection. In other words, in this case, energy and structure data are saved once every 10 steps. If you want to collect energy and structure data step by step, manually set up as follows:
[option]
energy_step_flag = True
struc_step_flag = True
| Name | Value | Default | Description |
|---|---|---|---|
check_mindist_opt | bool | True | If True, a mindist constraint is checked after structure relaxation. |
stop_chkpt | int | 0 | CrySPY stops at a specified check point. |
load_struc_flag | bool | False | If True, load initial structures from ./data/pkl_data/init_struc_data.pkl. |
stop_next_struc | bool | False | If True, CrySPY does not submit jobs for next structures, but jobs for next stage are submitted. |
recalc | int [int …] | (empty list) | Specify structure IDs if you want to recalculate or continue optimization. |
append_struc_ea | bool | False | If True, append structures by EA. |
energy_step_flag | bool | False | If True, save energy_step_data in ./data/pkl_data/energy_step_data.pkl. |
struc_step_flag | bool | False | If True, save struc_step_data in ./data/pkl_data/struc_step_data.pkl. |
force_step_flag | bool | False | If True, save force_step_data in ./data/pkl_data/force_step_data.pkl. |
stress_step_flag | bool | False | If True, save stress_step_data in ./data/pkl_data/stress_step_data.pkl. |
2024 April 22
CrySPY automatically generates the k-point setting using the pymatgen.io.vasp.Kpoints.automatic_density_by_vol function from pymatgen.
An example in cryspy.in with nstage = 2 is as follows:
[VASP]
kppvol = 40 120
kppvol means a grid density per Å
${}^{-3} $ of reciprocal cell.
VASP: gamma centered meshes are used for hexagonal cells and face-centered cells; otherwise, Monkhorst-Pack grids are employed.
QE and OMX: only a k-mesh is provided, no offset.
Here are the guidelines. We use VESTA for visualizing crystal structures.
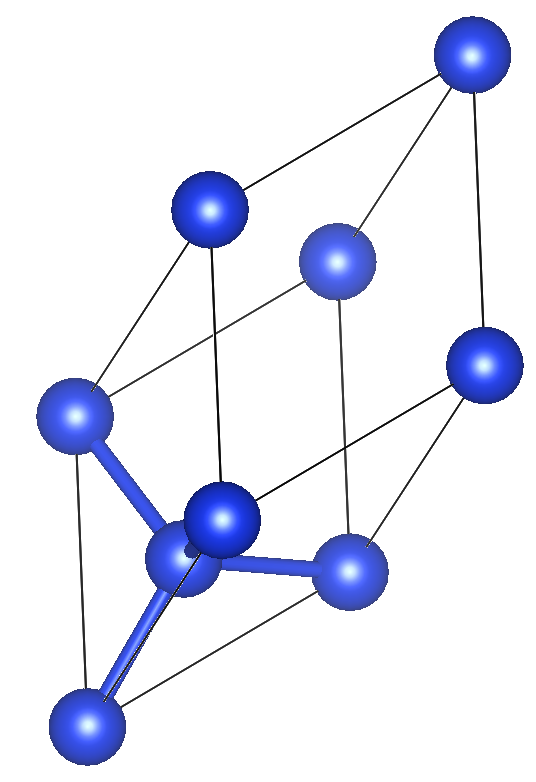
a = b = c = 3.836 Å
| kppvol | k-mesh |
|---|---|
| 0 | [1, 1, 1] |
| 20 | [4, 4, 4] |
| 40 | [6, 6, 6] |
| 60 | [7, 7, 7] |
| 80 | [7, 7, 7] |
| 100 | [8, 8, 8] |
| 120 | [9, 9, 9] |
| 140 | [9, 9, 9] |
| 160 | [9, 9, 9] |
| 180 | [10, 10, 10] |
| 200 | [10, 10, 10] |
| 400 | [13, 13, 13] |
| 600 | [15, 15, 15] |
| 800 | [17, 17, 17] |
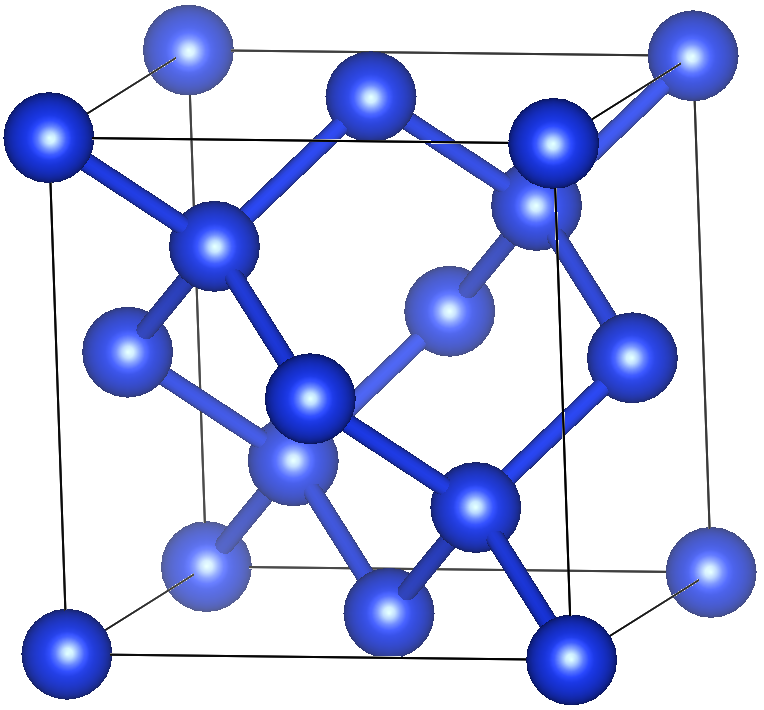
a = b = c = 5.431 Å
| kppvol | k-mesh |
|---|---|
| 0 | [1, 1, 1] |
| 20 | [3, 3, 3] |
| 40 | [3, 3, 3] |
| 60 | [4, 4, 4] |
| 80 | [4, 4 ,4] |
| 100 | [5, 5, 5] |
| 120 | [5, 5, 5] |
| 140 | [6, 6, 6] |
| 160 | [6, 6, 6] |
| 180 | [6, 6, 6] |
| 200 | [6, 6, 6] |
| 400 | [8, 8, 8] |
| 600 | [9, 9, 9] |
| 800 | [10, 10, 10] |
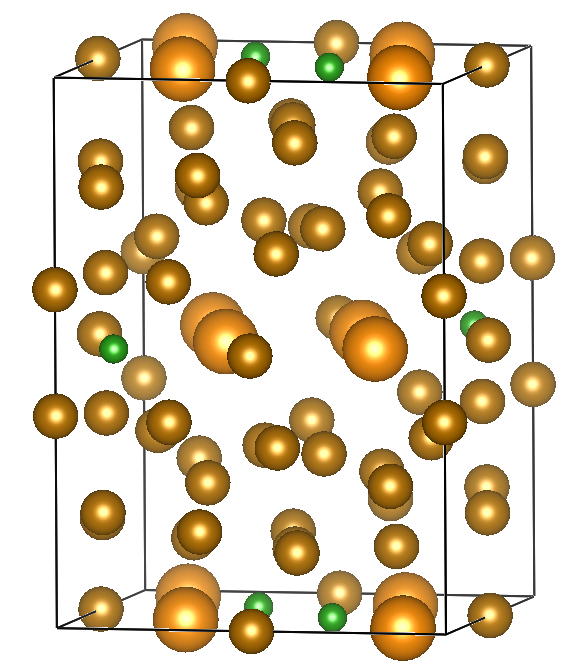
a = b = 8.804 Å
c = 12.205 Å
| kppvol | k-mesh |
|---|---|
| 0 | [1, 1, 1] |
| 20 | [1, 1, 1] |
| 40 | [2, 2, 1] |
| 60 | [2, 2, 2] |
| 80 | [3, 3 ,2] |
| 100 | [3, 3, 2] |
| 120 | [3, 3, 2] |
| 140 | [3, 3, 2] |
| 160 | [3, 3, 2] |
| 180 | [4, 4, 2] |
| 200 | [4, 4, 3] |
| 400 | [5, 5, 3] |
| 600 | [6, 6, 4] |
| 800 | [6, 6, 4] |