Input file
入力ファイルcryspy.inの解説.
入力ファイルcryspy.inの解説.
CrySPY uses the configparser module to read input file, cryspy.in .
cryspy.in consists of sections, led by a [section] header and followed by name = value or name : value entries.
Section names and values are case sensitive, but names are not.
Lines beginning with # or ; are ignored and may be used to provide comments.
Accepted bool values are 1, yes, true, and on, which cause this method to return True,
and 0, no, false, and off, which cause it to return False.
These string values for bool are checked in a case-insensitive manner.
Some values are given in a space-separated manner.
See configparser in detail.
section name: case sensitive
name: case insensitive
value: case sensitive except for bool
2025年3月6日 更新
| Name | Value | Default | Description |
|---|---|---|---|
algo | RS, EA, EA-vc, BO, LAQA | アルゴリズム | |
calc_code | VASP, QE, OMX, soiap, LAMMPS, ASE | 構造最適化の計算コード | |
tot_struc | int | 構造数.EAやEA-vcの場合は使用されない. | |
nstage | int | ステージ数 | |
njob | int | 同時に投入するジョブの数 | |
jobcmd | str | ジョブを投入するコマンド,例えばqsubやsbatch | |
jobfile | str | ジョブファイルのファイル名 |
2025年7月4日 更新
| Name | Value | Default | Description |
|---|---|---|---|
struc_mode | crystal, mol, mol_bs | crystal | 構造生成モード |
atype | atomic symbol [atomic symbol …] | Atom type. e.g. atype = Na Cl. EA-vcでは使用されない. | |
nat | int [int …] | atom typeに応じた原子数. e.g. nat = 8 8. | |
mindist (mindist_?) | float [float …] | None | 最小原子間距離の制限 [Å]. |
mindist_factor | float | 1.0 | mindistのスケーリング係数 |
vol_factor | float | 1.0 | 体積スケーリング係数 |
vol_mu | float | None | 体積の平均.体積を指定したい場合に使用 |
vol_sigma | float | None | 体積の標準偏差.体積を指定したい場合に使用 |
symprec | float | 0.01 | 対称性を見つける際の精度 |
spgnum | all, space group number, 0 | all | 空間群の制限.allであれば1–230の全て.0であれば空間群情報のない(対称性のない)ランダム構造生成 |
use_find_wy | bool | False | find_wyを使用して構造生成する際のオプション |
if algo is EA-vc
| Name | Value | Default | Description |
|---|---|---|---|
ll_nat | int [int …] | natの下限. e.g. ll_nat = 0 0. | |
ul_nat | int [int …] | natの上限. e.g. ul_nat = 8 8. | |
charge | int [int …] | None | 電荷中性条件を課したい時に使用.各atom typeの電荷. e.g. charge = 1 -1 for NaCl. |
if struc_mode is mol or mol_bs
| Name | Value | Default | Description |
|---|---|---|---|
mol_file | str [str …] | Path of molecule files or molecule names. | |
nmol | int [int …] | The number of molecules. | |
timeout_mol | float | None | Time out for molecular structure generation. |
rot_mol | random, random_mol, random_wyckoff | random_wyckoff | Only used in mol_bs. Mode for rotation of molecules. |
nrot | int | 20 | Only used in mol_bs. Maximum number of trials to rotate molecules. |
mindist_mol_bs (mindist_mol_bs_?) | float [float …] | None | Only used in mol_bs. Constraint on minimum intermolecular distance [Å]. |
mindist_mol_bs_factor | float | 1.0 | Only used in mol_bs. Scaling factor for mindist_mol_bs. |
if use_find_wy is True or spgnum = 0
| Name | Value | Default | Description |
|---|---|---|---|
fwpath | str | None | Only used with find_wy. Path of find_wy. If None, fwpath is automatically searched in your $PATH. |
minlen | float | Only used with find_wy or spgnum = 0. Minimum length of lattice vector [Å]. | |
maxlen | float | Only used with find_wy or spgnum = 0. Maximum length of lattice vector [Å]. | |
dangle | float | Only used with find_wy or spgnum = 0. Delta angle for alpha, beta, and gamma in degree unit. | |
maxcnt | int | 50 | Only used with find_wy or spgnum = 0. Maximum number of trials to determine atom positions. |
2025年7月12日 更新
VASPを使う場合は(calc_code = VASP)[VASP]セクションが必要になる.
| Name | Value | Default | Description |
|---|---|---|---|
kppvol | int [int …] | 各ステージにおける逆格子空間でのÅ**(-3)あたりのグリッド密度. | |
force_gamma | bool | False | Trueなら常にガンマ点を通るメッシュを使う. |
vasp_MAGMOM | float [float …] | None | EA-vcの時のみ使用.各元素に対応するMAGMOMの値. |
vasp_LDAUL | int [int …] | None | EA-vcの時のみ使用.各元素に対応するLDAULの値. |
vasp_LDAUU | float [float …] | None | EA-vcの時のみ使用.各元素に対応するLDAUUの値. |
vasp_LDAUJ | float [float …] | None | EA-vcの時のみ使用.各元素に対応するLDAUJの値. |
[QE] section is required only if you use QE (calc_code = QE)
| Name | Value | Default | Description |
|---|---|---|---|
kppvol | int [int …] | Grid density per Å**(-3) of reciprocal cell in each stage | |
qe_infile | str | File name of QE input file. | |
qe_outfile | str | File name of QE output file. | |
pv_term | bool | False | If true, read enthalpy instead of total energy. |
[OMX] section is required only if you use OpenMX (calc_code = OMX)
| Name | Value | Default | Description |
|---|---|---|---|
kppvol | int [int …] | Grid density per Å**(-3) of reciprocal cell in each stage | |
OMX_infile | str | File name of OpenMX input file. | |
OMX_outfile | str | File name of OpenMX output file. | |
ValenceElectrons | str float float [str float float …] | The number of initial charges for up and down spin states. |
e.g. in NaCl: ValenceElectrons = Na 4.5 4.5 Cl 3.5 3.5.
[soiap] section is required only if you use soiap (calc_code = soiap)
| Name | Value | Default | Description |
|---|---|---|---|
soiap_infile | str | File name of soiap input file. | |
soiap_outfile | str | File name of soiap output file. | |
soiap_cif | str | File name of soiap CIF-formatted initial structure. |
[LAMMPS] section is required only if you use LAMMPS (calc_code = LAMMPS)
| Name | Value | Default | Description |
|---|---|---|---|
lammps_infile | str | File name of LAMMPS input file. | |
lammps_outfile | str | File name of LAMMPS output file. | |
lammps_potential | str [str …], None | None | Potential. |
lammps_data | str | File name of LAMMPS data file. |
[ASE] section is required only if you use ASE (calc_code = ASE)
| Name | Value | Default | Description |
|---|---|---|---|
ase_python | str | File name of ASE input file. |
2025年7月7日 更新
| Name | Value | Default | Description |
|---|---|---|---|
n_pop | int | Population (see also Population size) | |
n_crsov | int | Number of offspring created by crossover | |
n_perm | int | Number of offspring created by permutation | |
n_strain | int | Number of offspring created by strain | |
n_rand | int | Number of structures created randomly | |
n_elite | int | Number of elite individuals (see also Natural selection) | |
fit_reverse | bool | False | If False, minimal search (see also Evaluate fitness) |
n_fittest | int | 0 | Number of individuals that remain natural selection. If set to 0, all individuals are retained. |
slct_func | TNM, RLT | Function to select parents | |
t_size | int | 3 | Tournament size. Used only used slct_func = TNM. (see also Tournament selection) |
a_rlt | float | 10.0 | Parameter for linear scaling. Used only with slct_func = RLT. (see also Roulette selection) |
b_rlt | float | 1.0 | Parameter for linear scaling. Used only with slct_func = RLT. (see also Roulette selection) |
crs_lat | equal, random | random | How to mix lattice vectors (see also crossover > 5. Swap the sliced halves) |
nat_diff_tole | int | 4 | Tolerance for difference in the number of atoms in crossover. (see also crossover > 6. Select the offspring with more atoms) |
ntimes | int | 1 | Number of times in permutation. |
sigma_st | float | 0.5 | Standard deviation for strain. |
maxcnt_ea | int | 50 | Maximum number of trials in EA. |
maxgen_ea | int | 0 | Maximum generation. If set to 0, no upper limit is applied. |
emax_ea | float | None | Energy upper limit (eV/atom) for natural selection. |
emin_ea | float | None | Energy lower limit (eV/atom) for natural selection. |
emax_ea and emin_ea are used not in natural selection but when computing the convex hull.| Name | Value | Default | Description |
|---|---|---|---|
n_add | int | Number of offspring created by addition. | |
add_max | int | 3 | (since version 1.4.1) Maximum number of atoms to add in addition |
n_elim | int | Number of offspring created by elimination. | |
elim_max | int | 3 | (since version 1.4.1) Maximum number of atoms to eliminate in elimination |
n_subs | int | Number of offspring created by substitution. | |
target | str | random | Target. Only random for now. |
end_point | (float, …, float) | Energy of end points for formation energy. | |
emax_ea | float | None | Energy upper limit (eV/atom) for computing the convex hull. |
emin_ea | float | None | Energy lower limit (eV/atom) for computing the convex hull. |
cgen | int | None | (since version 1.4.1) Which generation’s data to plot up to. If None, data will be plotted up to the latest generation. |
show_max | float | 0.2 | When plotting the convex hull, the maximum value of the y-axis (for binary systems) or the maximum hull distance (for ternary systems) is set by show_max. |
lable_stable | bool | True | Whether to show stable compositions when plotting the convex hull. |
vmax | float | 0.2 | Maximum value of the colorbar representing hull distance. |
bottom_margin | float | 0.02 | Bottom margin of the y-axis for binary convex hull plot. |
fig_format | str | svg | Figure format for convex hull plot: svg, png, or pdf. |
2024 May 27th, updated
[BO] section is required only if you use BO (algo = BO)
| Name | Value | Default | Description |
|---|---|---|---|
nselect_bo | int | The number of structures to be selected at once. | |
score | TS, EI, PI | Acquisition function. | |
num_rand_basis | int | 0 | If 0, Gaussian process. The number of basis function. |
cdev | float | 0.001 | Cutoff of deviation for standardization. |
dscrpt | FP | Structure descriptor. | |
max_select_bo | int | 0 | Maximum number of selection. |
manual_select_bo | int [int …] | None | Structure IDs to be selected manually. |
emax_bo | float | None | Upper limit of energy in BO. |
emin_bo | float | None | Lower limit of energy in BO. |
if decrpt is FP
CrySPY 1.3.0 or later
fppath and fp_rmin are obsolete.
| Name | Value | Default | Description |
|---|---|---|---|
fp_rmax | float | 8.0 | Only used with dscrpt = FP. Maximum cutoff of r in fingerprint. |
fp_npoints | int | 20 | Only used with dscrpt = FP. Number of discretized points for each pair in fingerprint. |
fp_sigma | float | 0.7 | Only used with dscrpt = FP. Sigma parameter [Å] in Gaussian smearing function. |
CrySPY 1.2.5 or earlyer
| Name | Value | Default | Description |
|---|---|---|---|
fppath | str | None | Only used with dscrpt = FP. Path of cal_fingerprint. If None, fwpath is automatically searched in your $PATH. |
fp_rmin | float | 0.5 | Only used with dscrpt = FP. Minimum cutoff of r in fingerprint. |
fp_rmax | float | 5.0 | Only used with dscrpt = FP. Maximum cutoff of r in fingerprint. |
fp_npoints | int | 20 | Only used with dscrpt = FP. Number of discretized points for each pair in fingerprint. |
fp_sigma | float | 1.0 | Only used with dscrpt = FP. Sigma parameter [Å] in Gaussian smearing function. |
LAQA (algo = LAQA)を使う場合,[LAQA]セクションが必要.
| Name | Value | Default | Description |
|---|---|---|---|
nselect_laqa | int | 1度の選択で選ばれる構造の数. | |
wf | float | 0.1 | 力の項の重み. |
ws | float | 10.0 | ストレス項の重み. |
See also Search algorithms > LAQA
algo = LAQAでは[option]セクションの下記の二つは自動的にTrueになる.
原子に働く力とストレスのデータは1ステップごとに収集される. エネルギーと構造データは1ステップごとではなく,選択ごとに収集される. つまり,この場合は10ステップおきにエネルギーと構造データは保存される. もし1ステップごとのデータが欲しいのであれば,手動で下記の設定を追加すること.
[option]
energy_step_flag = True
struc_step_flag = True
| Name | Value | Default | Description |
|---|---|---|---|
check_mindist_opt | bool | True | If True, a mindist constraint is checked after structure relaxation. |
stop_chkpt | int | 0 | CrySPY stops at a specified check point. |
load_struc_flag | bool | False | If True, load initial structures from ./data/pkl_data/init_struc_data.pkl. |
stop_next_struc | bool | False | If True, CrySPY does not submit jobs for next structures, but jobs for next stage are submitted. |
recalc | int [int …] | (empty list) | Specify structure IDs if you want to recalculate or continue optimization. |
append_struc_ea | bool | False | If True, append structures by EA. |
energy_step_flag | bool | False | If True, save energy_step_data in ./data/pkl_data/energy_step_data.pkl. |
struc_step_flag | bool | False | If True, save struc_step_data in ./data/pkl_data/struc_step_data.pkl. |
force_step_flag | bool | False | If True, save force_step_data in ./data/pkl_data/force_step_data.pkl. |
stress_step_flag | bool | False | If True, save stress_step_data in ./data/pkl_data/stress_step_data.pkl. |
2024年4月22日
CrySPYではk-pointのセッティングはpymatgenのpymatgen.io.vasp.Kpoints.automatic_density_by_vol関数を使って自動生成している.
以下はcryspy.inでnstage = 2の場合の例:
[VASP]
kppvol = 40 120
kppvolは逆格子空間でのÅ
${}^{-3} $あたりのグリッド密度.
VASP:hexagonalとface-centeredセルではガンマ点を通るメッシュが使われ,それ以外ではMonkhorst-Packが使われる.
QE and OMX:kメッシュだけ利用,オフセットは設定しない.
下記を参考に.結晶構造の画像はVESTAを使用.
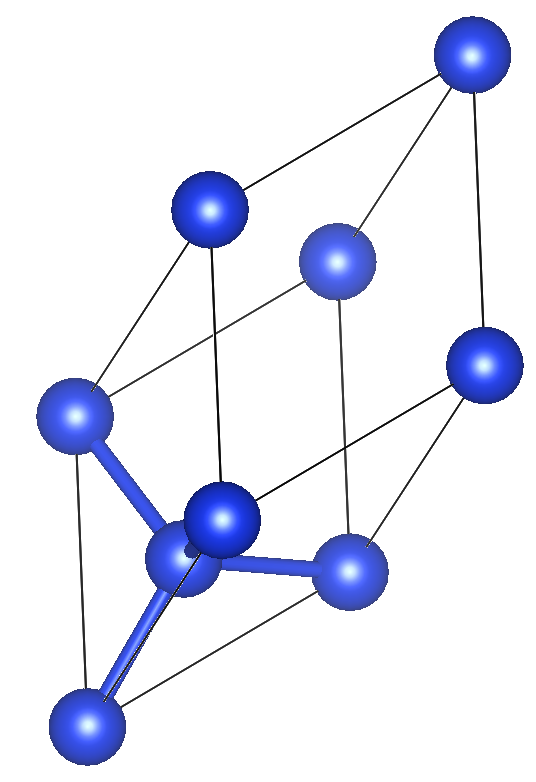
a = b = c = 3.836 Å
| kppvol | k-mesh |
|---|---|
| 0 | [1, 1, 1] |
| 20 | [4, 4, 4] |
| 40 | [6, 6, 6] |
| 60 | [7, 7, 7] |
| 80 | [7, 7, 7] |
| 100 | [8, 8, 8] |
| 120 | [9, 9, 9] |
| 140 | [9, 9, 9] |
| 160 | [9, 9, 9] |
| 180 | [10, 10, 10] |
| 200 | [10, 10, 10] |
| 400 | [13, 13, 13] |
| 600 | [15, 15, 15] |
| 800 | [17, 17, 17] |
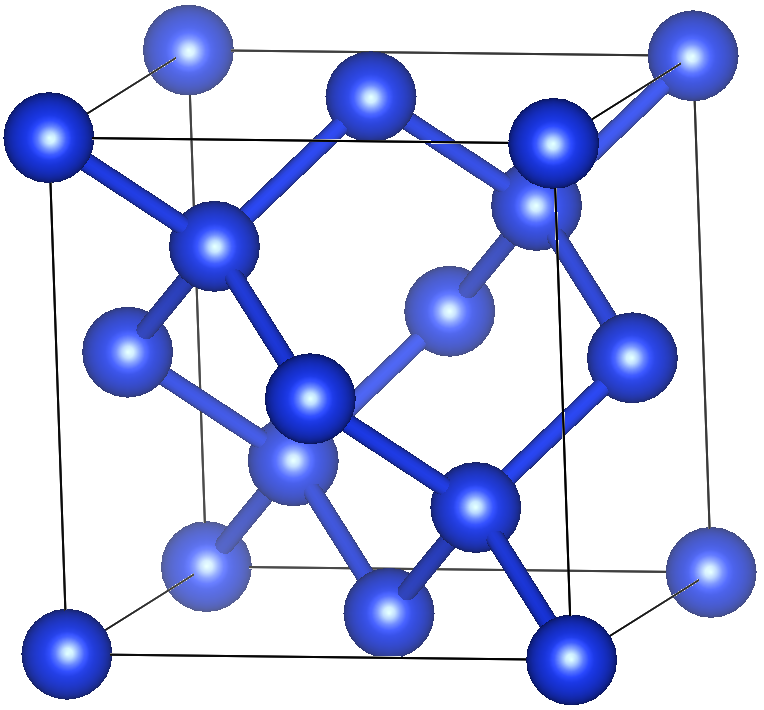
a = b = c = 5.431 Å
| kppvol | k-mesh |
|---|---|
| 0 | [1, 1, 1] |
| 20 | [3, 3, 3] |
| 40 | [3, 3, 3] |
| 60 | [4, 4, 4] |
| 80 | [4, 4 ,4] |
| 100 | [5, 5, 5] |
| 120 | [5, 5, 5] |
| 140 | [6, 6, 6] |
| 160 | [6, 6, 6] |
| 180 | [6, 6, 6] |
| 200 | [6, 6, 6] |
| 400 | [8, 8, 8] |
| 600 | [9, 9, 9] |
| 800 | [10, 10, 10] |
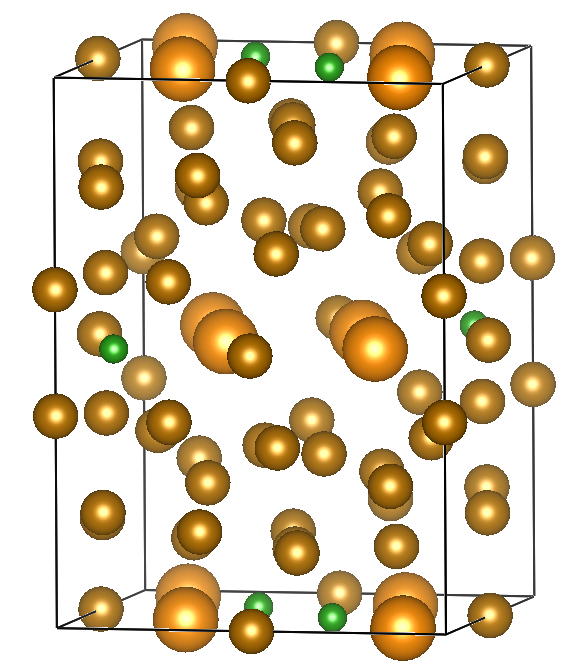
a = b = 8.804 Å
c = 12.205 Å
| kppvol | k-mesh |
|---|---|
| 0 | [1, 1, 1] |
| 20 | [1, 1, 1] |
| 40 | [2, 2, 1] |
| 60 | [2, 2, 2] |
| 80 | [3, 3 ,2] |
| 100 | [3, 3, 2] |
| 120 | [3, 3, 2] |
| 140 | [3, 3, 2] |
| 160 | [3, 3, 2] |
| 180 | [4, 4, 2] |
| 200 | [4, 4, 3] |
| 400 | [5, 5, 3] |
| 600 | [6, 6, 4] |
| 800 | [6, 6, 4] |