チュートリアル
情報
初心者の方はランダムサーチから始めると良い.Exampleファイルはこちら –> cryspy_utility.
初心者の方はランダムサーチから始めると良い.Exampleファイルはこちら –> cryspy_utility.
ASE は初心者にとって始めやすいツールである. CrySPYのインストール時に,ASEも自動的にインストールされるためである. 高い精度はないが,非常に軽量で高速な原子間ポテンシャルを備えており,ノートPCなどの低スペックな環境でも動作確認に適している.
下記のどれか一つに従い,その後CrySPY実行のセクションに移ること.
2025年7月11日 更新 v1.4.2
ASEは様々なコードのインターフェースを提供しているPythonライブラリであり, Pure Python EMT calculatorというシンプルなEMTの計算も実行できる.CrySPYさえインストールしてあれば,精度はともかく簡単に計算できるので,CrySPYのテストにちょうど良い.
このチュートリアルでは,MacやLinuxなどのOSのローカルPCを用いてCu 8原子の構造探索を試す.
ここでは次のような条件を想定している:
job_cryspyase_in.pyどこか適当なワーキングディレクトリに移動して,まずはexampleをコピーしてくる.下記のどちらからコピーしてきても良い.
cd ase_Cu8_RS
tree
.
├── calc_in
│ ├── ase_in.py
│ └── job_cryspy
└── cryspy.in
cryspy.inはCrySPYの入力ファイル.
[basic]
algo = RS
calc_code = ASE
tot_struc = 5
nstage = 1
njob = 5
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Cu
nat = 8
[ASE]
ase_python = ase_in.py
[option]
[basic] セクションのjobcmd = zshは環境に合わせてjobcmd = shやjobcmd = bash等に変更する.
CrySPYは内部でバックグラウンドジョブとしてzsh job_cryspyを実行する.
ASEを使う場合は,[ASE]セクションが必要.
下記の二つのファイル名は好きなように変えても良い.
jobfile: job_cryspyase_python: ase_in.py他の入力変数については後で説明を行う.
ASEのジョブファイルや入力ファイルはこのディレクトリに準備する.
ジョブファイルの名前はcryspy.inのjobfileに一致させる必要がある.
ジョブファイルの例は下記の通り.
#!/bin/sh
# ---------- ASE
python3 ase_in.py
ase_in.pyというファイル名も自由に変えられるが, cryspy.inのase_pythonの値と一致させておく必要がある.
CrySPYではジョブファイルの最後の行はsed -i -e ‘3 s/^.*$/done/’ stat_jobとしておくルールになっている.
バージョン1.4.2からCrySPYがジョブファイルの末尾に自動的に下記を追記するようになった.(参考:機能 > ジョブファイルの自動書き換え)
# ---------- CrySPY
sed -i -e '3s/^sub.*/done/' stat_job
1.4.2より古いバージョンでは,ジョブファイルの最後の行はsed -i -e '3s/^sub.*/done/' stat_jobと書いておくルールになっている.自分でsedコマンド文を書いたジョブファイルを1.4.2以上のバージョンで使用しても2回実行されるだけなので問題はない.
上記sedコマンドの意味は,stat_jobというファイルの3行目のsubから始まる部分をdoneに変える処理.
(詳細:機能 > ジョブファイルの自動書き換え)
CrySPYのジョブファイルのCrySPY_IDという文字列は自動的に構造IDに置き換わるようになっている.
PBSやSLURMといったジョブスケジューラーを使う場合,ジョブ名にCrySPY_IDと書いておくとどの構造のジョブなのかが分かり便利である.
例えば,PBSでは#PBS -N Si_CrySPY_IDのように書いておくと,ジョブをサブミットする際,#PBS -N Si_10のように置き換わる.
注意点として,ジョブ名を数字から始めるとエラーとなることが多いので,Si_のように何か文字列を頭につけておくこと.
ステージ数(nstage in cryspy.in)に応じた数のインプットファイルが必要となる.
インプットファイル名の先頭にx_,または語尾に_xをつけて準備する.
ここでxはステージ数.
CrySPYが探すインプットファイル名の優先順位は以下の通り.
x_ase_in.pyase_in.py_xase_in.py各ステージで共通のインプットを使用するのであればx_や_xを省略できる.
このASEのチュートリアルではnstage = 1を用いるので,ASEのインプットファイルはase_in.pyの一つだけでよい.x_や_xを省略したファイルを準備している.
ase_in.pyは例えば下記の通り(ASEの使い方の詳細は公式のドキュメントを見ること).
from ase.constraints import FixSymmetry
from ase.filters import FrechetCellFilter
from ase.calculators.emt import EMT
from ase.optimize import BFGS
from ase.io import read, write
# ---------- input structure
# CrySPY outputs 'POSCAR' as an input file in work/xxxxxx directory
atoms = read('POSCAR', format='vasp')
# ---------- setting and run
atoms.calc = EMT()
atoms.set_constraint([FixSymmetry(atoms)])
cell_filter = FrechetCellFilter(atoms, hydrostatic_strain=False)
opt = BFGS(cell_filter)
# ---------- run
converged = opt.run(fmax=0.01, steps=2000)
# ---------- rule in ASE interface
# output file for energy: 'log.tote' in eV/cell
# CrySPY reads the last line of 'log.tote' file
# outimized structure: 'CONTCAR' file in vasp format
# check_opt: 'out_check_opt' file ('done' or 'not yet')
# CrySPY reads the last line of 'out_check_opt' file
# ------ energy
e = cell_filter.atoms.get_total_energy() # eV/cell
with open('log.tote', mode='w') as f:
f.write(str(e))
# ------ struc
opt_atoms = cell_filter.atoms.copy()
opt_atoms.set_constraint(None) # remove constraint for pymatgen
write('CONTCAR', opt_atoms, format='vasp', direct=True)
# ------ check_opt
with open('out_check_opt', mode='w') as f:
if converged:
f.write('done\n')
else:
f.write('not yet\n')
ASEはVASPやQEなどと違って,入力ファイル(python script)は自分で書くことになるので自由度がある. CrySPYでは3つのルールを設けている.
log.toteというファイルに出力する.CrySPYはこのファイルの最後の行を読む.CONTCARというファイルにVASPフォーマットで出力する.out_check_optというファイルにdoneかnot_yetで書き込む.CrySPYはこのファイルの最後の行を読む.ここまで準備ができたらCrySPY実行へ進む.
ここに書いてある内容は少し古いので,最近のチュートリアルはチュートリアル > ランダムサーチ(RS) > ASE on your local PCを見ること.このページの内容も動作には問題ないはず.
2025年3月6日 更新
soiapは原子間ポテンシャルを使用した計算ができるソフトウェアであり,計算が軽いのでCrySPYのテストにちょうど良い. soiapのインストールや詳細はinstructionsを参照.
このチュートリアルでは,MacやLinuxのローカルPC上でCrySPYを試す. テストシステムはSi 8原子.
ここでは次のような条件を想定している:
~/CrySPY_root/CrySPY-0.9.0/cryspy.pyjob_cryspy~/local/soiap-0.3.0/src/soiapsoiap.insoiap.outinitial.cifどこか適当なワーキングディレクトリに移動して,まずはexampleをコピーしてくる.下記のどちらからコピーしてきても良い.
cp -r ~/CrySPY_root/CrySPY-0.9.0/example/v0.9.0/soiap_RS_Si8 .cd soiap_RS_Si8
tree
.
├── calc_in
│ ├── job_cryspy
│ └── soiap.in_1
└── cryspy.in
cryspy.inがCrySPYの入力ファイル.
[basic]
algo = RS
calc_code = soiap
tot_struc = 5
nstage = 1
njob = 2
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Si
nat = 8
[soiap]
soiap_infile = soiap.in
soiap_outfile = soiap.out
soiap_cif = initial.cif
[option]
[basic] セクションのjobcmd = zshは環境に合わせてjobcmd = shやjobcmd = bash等に変更する. CrySPYは内部でバックグラウンドジョブとしてzsh job_cryspyを実行する.
soiapを使う場合は[soiap]セクションが必要となる.下記のファイル名は好きなように変えても良い.
jobfilesoiap_infilesoiap_outfilesoiap_cif他の入力変数については後で説明を行う.
soiapのジョブファイルや入力ファイルはこのディレクトリに準備する.
ジョブファイルの名前はcryspy.inのjobfileに一致させる必要がある.
ジョブファイルの例は下記の通り.
#!/bin/sh
# ---------- soiap
EXEPATH=/path/to/soiap
$EXEPATH/soiap soiap.in 2>&1 > soiap.out
# ---------- CrySPY
sed -i -e '3 s/^.*$/done/' stat_job
/path/to/soiapはsoiapの実行ファイルのpathに変えること.
入力ファイル(soiap.in)と出力ファイル(soiap.out)はcryspy.inで指定したsoiap_infileとsoiap_outfileに合わせること.
最後の行以外は普段使っているようなジョブスクリプトで良いが,
CrySPYではジョブファイルの最後の行はsed -i -e '3 s/^.*$/done/' stat_jobとしておくルールになっている.
ジョブファイルの最後の行はsed -i -e '3 s/^.*$/done/' stat_jobと書いておく.
CrySPYのジョブファイルのCrySPY_IDという文字列は自動的に構造IDに置き換わるようになっている.
PBSやSLURMといったジョブスケジューラーを使う場合,ジョブ名にCrySPY_IDと書いておくとどの構造のジョブなのかが分かり便利である.
例えば,PBSでは#PBS -N Si_CrySPY_IDのように書いておくと,ジョブをサブミットする際,#PBS -N Si_10のように置き換わる.
注意点として,ジョブ名を数字から始めるとエラーとなることが多いので,Si_のように何か文字列を頭につけておくこと.
ステージ数(nstage in cryspy.in)に応じた数のインプットファイルが必要となる.
インプットファイル名の語尾に_xをつけて準備する.
ここでxはステージ数.
soiapのチュートリアルではnstage = 1を用いるので,インプットファイルはsoiap.in_1の一つだけが必要.
soiap.in_1は例えば下記の通り.
crystal initial.cif ! CIF file for the initial structure
symmetry 1 ! 0: not symmetrize displacements of the atoms or 1: symmetrize
md_mode_cell 3 ! cell-relaxation method
! 0: FIRE, 2: quenched MD, or 3: RFC5
number_max_relax_cell 100 ! max. number of the cell relaxation
number_max_relax 1 ! max. number of the atom relaxation
max_displacement 0.1 ! max. displacement of atoms in Bohr
external_stress_v 0.0 0.0 0.0 ! external pressure in GPa
th_force 5d-5 ! convergence threshold for the force in Hartree a.u.
th_stress 5d-7 ! convergence threshold for the stress in Hartree a.u.
force_field 1 ! force field
! 1: Stillinger-Weber for Si, 2: Tsuneyuki potential for SiO2,
! 3: ZRL for Si-O-N-H, 4: ADP for Nd-Fe-B, 5: Jmatgen, or
! 6: Lennard-Jones
1行目に書く初期構造ファイル(initial.cif)はcryspy.inのsoiap_cifの値と揃える.
ここまで準備ができたらCrySPY実行へ進む.
2025年7月12日 更新
このチュートリアルでは,PBSなどのジョブスケジューラーを備えたPCクラスターを想定してCrySPYを試す.第一原理計算のVASPを用いて,Na8Cl8(16原子)の構造探索を行う.
ここでは次のような条件を想定している:
qsubjob_cryspyどこか適当なワーキングディレクトリに移動して,まずはexampleをコピーしてくる.下記のどちらからコピーしてきても良い.
cd vasp_Na8Cl8_RS
tree
.
├── calc_in
│ ├── 1_INCAR
│ ├── 2_INCAR
│ ├── POTCAR_dummy
│ └── job_cryspy
└── cryspy.in
cryspy.inはCrySPYの入力ファイル.
[basic]
algo = RS
calc_code = VASP
tot_struc = 5
nstage = 2
njob = 2
jobcmd = qsub
jobfile = job_cryspy
[structure]
atype = Na Cl
nat = 8 8
mindist_1 = 2.5 1.5
mindist_2 = 1.5 2.5
[VASP]
kppvol = 40 80
[option]
[basic] セクションのjobcmd = qsubは環境に合わせて変更する.
CrySPYは内部でバックグラウンドジョブとしてqsub job_cryspyを実行する.
下記のファイル名は好きなように変えても良い.
jobfile構造最適化計算はステージ制を採用しており,ここではnstage = 2を用いている.
例えば,最初のステージでは,セルを固定し内部座標だけ緩和する設定で,k点も少ない計算を実行し,2ステージ目でセルも含めてフルに構造緩和して,精度も高めるようなことが可能となっている.
VASPを使う場合は,[VASP]セクションが必要.
ここでは各ステージにおけるk点のグリッド密度(Å^-3)をkppvolに指定する必要がある.
kppvolの詳細はこちら –> Input file > Kpoint
他のインプット変数に関しては後ほど説明する.
ジョブファイルやVASPのインプットをこのディレクトリに置く.
ジョブファイルの名前はcryspy.inのjobfileに一致させる必要がある.
ジョブファイルの例は下記の通り.
#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
####$ -V -S /bin/zsh
#$ -N Na8Cl8_CrySPY_ID
#$ -pe smp 20
####$ -q ibis1.q
####$ -q ibis2.q
####$ -q ibis3.q
####$ -q ibis4.q
# ---------- vasp
VASPROOT=/usr/local/vasp/vasp.6.4.2/bin
mpirun -np $NSLOTS $VASPROOT/vasp_std
VASPROOTは環境に合わせて変更する.普段VASPのジョブを流しているジョブファイルを使えば良い.
CrySPYではジョブファイルの最後の行はsed -i -e ‘3 s/^.*$/done/’ stat_jobとしておくルールになっている.
バージョン1.4.2からCrySPYがジョブファイルの末尾に自動的に下記を追記するようになった.(参考:機能 > ジョブファイルの自動書き換え)
# ---------- CrySPY
sed -i -e '3s/^sub.*/done/' stat_job
1.4.2より古いバージョンでは,ジョブファイルの最後の行はsed -i -e '3s/^sub.*/done/' stat_jobと書いておくルールになっている.自分でsedコマンド文を書いたジョブファイルを1.4.2以上のバージョンで使用しても2回実行されるだけなので問題はない.
上記sedコマンドの意味は,stat_jobというファイルの3行目のsubから始まる部分をdoneに変える処理.
(詳細:機能 > ジョブファイルの自動書き換え)
CrySPYのジョブファイルのCrySPY_IDという文字列は自動的に構造IDに置き換わるようになっている.
PBSやSLURMといったジョブスケジューラーを使う場合,ジョブ名にCrySPY_IDと書いておくとどの構造のジョブなのかが分かり便利である.
例えば,PBSでは#PBS -N Si_CrySPY_IDのように書いておくと,ジョブをサブミットする際,#PBS -N Si_10のように置き換わる.
注意点として,ジョブ名を数字から始めるとエラーとなることが多いので,Si_のように何か文字列を頭につけておくこと.
ステージ数(nstage in cryspy.in)に応じた数のインプットファイルが必要となる.
インプットファイル名の先頭にx_,または語尾に_xをつけて準備する.
ここでxはステージ数.
CrySPYが探すインプットファイル名の優先順位は以下の通り.
x_INCARINCAR_xINCAR各ステージで共通のインプットを使用するのであればx_や_xを省略できる.
今はnstage = 2を用いているので,1_INCAR_1と2_INCARが必要となる.
ここでは,1_INCARはセルを固定して内部座標だけ緩和する設定,2_INCARはセルも含めてフルに緩和する設定になっている.
1_INCAR
SYSTEM = NaCl
!!!LREAL = Auto
Algo = Fast
NSW = 40
LWAVE = .FALSE.
!LCHARG = .FALSE.
ISPIN = 1
ISMEAR = 0
SIGMA = 0.1
IBRION = 2
ISIF = 2
EDIFF = 1e-5
EDIFFG = -0.01
2_INCAR
SYSTEM = NaCl
!!LREAL = Auto
Algo = Fast
NSW = 200
ENCUT = 341
!!LWAVE = .FALSE.
!!LCHARG = .FALSE.
ISPIN = 1
ISMEAR = 0
SIGMA = 0.1
IBRION = 2
ISIF = 3
EDIFF = 1e-5
EDIFFG = -0.01
CrySPYはPOSCARとKPOINTSファイルを自動生成する.
POTCARファイルはユーザーが準備する必要がある.
このexampleに含まれているPOTCARは空のファイルなので,各自で準備すること.
exampleに含まれているPOTCARは空のファイル.配布できない.
ここまで準備ができたらCrySPY実行に進む.
2025年7月18日 更新
このチュートリアルでは,PBSなどのジョブスケジューラーを備えたPCクラスターを想定してCrySPYを試す.第一原理計算のQUANTUM ESPRESSOを用いて,Si 8原子の構造探索を行う.
ここでは次のような条件を想定している:
qsubjob_cryspy/usr/local/qe-6.5/bin/pw.xpwscf.inpwscf.outどこか適当なワーキングディレクトリに移動して,まずはexampleをコピーしてくる.下記のどちらからコピーしてきても良い.
cd QE_RS_Si8
tree
.
├── calc_in
│ ├── job_cryspy
│ ├── 1_pwscf.in
│ └── 2_pwscf.in
└── cryspy.in
cryspy.inはCrySPYの入力ファイル.
[basic]
algo = RS
calc_code = QE
tot_struc = 5
nstage = 2
njob = 2
jobcmd = qsub
jobfile = job_cryspy
[structure]
atype = Si
nat = 8
[QE]
qe_infile = pwscf.in
qe_outfile = pwscf.out
kppvol = 40 80
[option]
[basic] セクションのjobcmd = qsubは環境に合わせて変更する.
CrySPYは内部でバックグラウンドジョブとしてqsub job_cryspyを実行する.
構造最適化計算はステージ制を採用しており,ここではnstage = 2を用いている.
例えば,最初のステージでは,セルを固定し内部座標だけ緩和する設定で,k点も少ない計算を実行し,2ステージ目でセルも含めてフルに構造緩和して,精度も高めるようなことが可能となっている.
QEを使う場合は,[QE]セクションが必要.
ここでは各ステージにおけるk点のグリッド密度(Å^-3)をkppvolに指定する必要がある.
kppvolの詳細はこちら –> Input file > Kpoint
下記のファイル名は好きなように変えても良い.
jobfileqe_infileqe_outfile他のインプット変数に関しては後ほど説明する.
ジョブファイルやQEのインプットをこのディレクトリに置く.
ジョブファイルの名前はcryspy.inのjobfileに一致させる必要がある.
ジョブファイルの例は下記の通り.
#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
####$ -V -S /bin/zsh
#$ -N Si8_CrySPY_ID
#$ -pe smp 20
####$ -q ibis1.q
####$ -q ibis2.q
mpirun -np $NSLOTS /path/to/pw.x < pwscf.in > pwscf.out
if [ -e "CRASH" ]; then
sed -i -e '3 s/^.*$/skip/' stat_job
exit 1
fi
/path/to/pw.xは環境に合わせて変更する.
入力(pwscf.in)出力(pwscf.out)ファイルの名前は好きに変えて良いが,cryspy.inのqe_infileとqe_outfileに合わせる必要がある.
普段QEのジョブを流しているジョブファイルを使えば良い.
CrySPYではジョブファイルの最後の行はsed -i -e ‘3 s/^.*$/done/’ stat_jobとしておくルールになっている.
バージョン1.4.2からCrySPYがジョブファイルの末尾に自動的に下記を追記するようになった.(参考:機能 > ジョブファイルの自動書き換え)
# ---------- CrySPY
sed -i -e '3s/^sub.*/done/' stat_job
1.4.2より古いバージョンでは,ジョブファイルの最後の行はsed -i -e '3s/^sub.*/done/' stat_jobと書いておくルールになっている.自分でsedコマンド文を書いたジョブファイルを1.4.2以上のバージョンで使用しても2回実行されるだけなので問題はない.
上記sedコマンドの意味は,stat_jobというファイルの3行目のsubから始まる部分をdoneに変える処理.
(詳細:機能 > ジョブファイルの自動書き換え)
CrySPYのジョブファイルのCrySPY_IDという文字列は自動的に構造IDに置き換わるようになっている.
PBSやSLURMといったジョブスケジューラーを使う場合,ジョブ名にCrySPY_IDと書いておくとどの構造のジョブなのかが分かり便利である.
例えば,PBSでは#PBS -N Si_CrySPY_IDのように書いておくと,ジョブをサブミットする際,#PBS -N Si_10のように置き換わる.
注意点として,ジョブ名を数字から始めるとエラーとなることが多いので,Si_のように何か文字列を頭につけておくこと.
ステージ数(nstage in cryspy.in)に応じた数のインプットファイルが必要となる.
インプットファイル名の先頭にx_,または語尾に_xをつけて準備する.
ここでxはステージ数.
CrySPYが探すインプットファイル名の優先順位は以下の通り.
x_pwscf.inpwscf.in_xpwscf.in各ステージで共通のインプットを使用するのであればx_や_xを省略できる.
今はnstage = 2を用いているので,1_pwscf.inと2_pwscf.inが必要となる.
ここでは,1_pwscf.inはセルを固定して内部座標だけ緩和する設定,2_pwscf.inはセルも含めてフルに緩和する設定になっている.
1_pwscf.in
&control
title = 'Si8'
calculation = 'relax'
nstep = 100
restart_mode = 'from_scratch',
pseudo_dir = '/usr/local/pslibrary.1.0.0/pbe/PSEUDOPOTENTIALS/'
outdir='./out.d/'
/
&system
ibrav = 0
nat = 8
ntyp = 1
ecutwfc = 44.0
occupations = 'smearing'
degauss = 0.01
/
&electrons
/
&ions
/
&cell
/
ATOMIC_SPECIES
Si 28.086 Si.pbe-n-kjpaw_psl.1.0.0.UPF
2_pwscf.in
&control
title = 'Si8'
calculation = 'vc-relax'
nstep = 200
restart_mode = 'from_scratch',
pseudo_dir = '/usr/local/pslibrary.1.0.0/pbe/PSEUDOPOTENTIALS/'
outdir='./out.d/'
/
&system
ibrav = 0
nat = 8
ntyp = 1
ecutwfc = 44.0
occupations = 'smearing'
degauss = 0.01
/
&electrons
/
&ions
/
&cell
/
ATOMIC_SPECIES
Si 28.086 Si.pbe-n-kjpaw_psl.1.0.0.UPF
pseudo_dirは各自の環境に合わせて変更する.
Inputs for structure data and k-point such as インプットファイルのATOMIC_POSITIONSとK_POINTSはCrySPYがpymatgenを用いて自動生成するのでユーザーが書く必要はない.
ここまで準備ができたらCrySPY実行に進む.
Coming soon.
Coming soon.
2025年6月16日 更新
詳細は 入力ファイルのページも見ること.
cryspy.inをもう一度チェックしてみよう.ここでの例は選んだcalc_codeに応じて,準備したものと少し異なるかもしれない.
[basic]
algo = RS
calc_code = ASE
tot_struc = 5
nstage = 1
njob = 5
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Cu
nat = 8
[ASE]
ase_python = ase_in.py
[option]
algo: アルゴリズム.ランダムサーチの場合はRSを使う.calc_code: 構造最適化のコード. VASP, QE, OMX, soiap, LAMMPS, ASEから選択.tot_struc: 構造数.この場合初回実行で5構造ランダムに生成される.nstage: ステージ数njob: 同時にサブミットするジョブの数.この例では2つのスロットを設定,言い換えると,2構造ずつ最適化を行う.jobcmd: ジョブを実行または投入するコマンド. bash, zsh, qsubなどjobfile: ジョブファイルのファイル名atype: Atom type. Na8Cl8の例: atype = Na Cl.nat: atypeに応じた原子数. Na8Cl8の例: nat = 8 8For version 1.0.0 or later, skip this page. The executable script is automatically installed.
Here, we assume the following condition:
~/CrySPY_root/CrySPY-0.9.0/cryspy.pyLet’s make a convenient shell script to avoid typing long commands over and over again.
Here, we create the script, cryspy (any file name will do).
$ emacs cryspy
$ chmod 744 cryspy
$ cat cryspy
#!/bin/sh
python3 -u ~/CrySPY_root/CrySPY-0.9.0/cryspy.py 1>> log 2>> err
-u option (unbuffered option) can be omitted.
You can put this script in your $PATH, or just use like bash ./cryspy.
2025 March 6, updated
Make sure you have the following in your working directory.
$ ls
calc_in/ cryspy.in
Then, run CyrSPY!
cryspy
If you use old version (0.10.3 or earlier):
bash ./cryspy
At the first run, CrySPY goes into structure generation mode. CrySPY stops after 5 structure generation.
If it worked properly, the following output appears on the screen:
[2025-03-06 18:52:21,495][cryspy_init][INFO]
Start CrySPY 1.4.0
[2025-03-06 18:52:21,495][cryspy_init][INFO] # ---------- Library version info
[2025-03-06 18:52:21,495][cryspy_init][INFO] pandas version: 2.2.2
[2025-03-06 18:52:21,495][cryspy_init][INFO] pymatgen version: 2025.1.24
[2025-03-06 18:52:21,495][cryspy_init][INFO] pyxtal version: 1.0.6
[2025-03-06 18:52:21,495][cryspy_init][INFO] # ---------- Read input file, cryspy.in
[2025-03-06 18:52:21,496][write_input][INFO] [basic]
[2025-03-06 18:52:21,496][write_input][INFO] algo = RS
[2025-03-06 18:52:21,496][write_input][INFO] calc_code = ASE
[2025-03-06 18:52:21,496][write_input][INFO] tot_struc = 5
[2025-03-06 18:52:21,496][write_input][INFO] nstage = 1
[2025-03-06 18:52:21,496][write_input][INFO] njob = 2
[2025-03-06 18:52:21,496][write_input][INFO] jobcmd = zsh
[2025-03-06 18:52:21,496][write_input][INFO] jobfile = job_cryspy
...
(omitted)
...
[2025-03-06 18:52:21,497][rs_gen][INFO] # ---------- Initial structure generation
[2025-03-06 18:52:21,497][rs_gen][INFO] # ------ mindist
[2025-03-06 18:52:21,498][struc_util][INFO] Cu - Cu: 1.32
[2025-03-06 18:52:21,498][rs_gen][INFO] # ------ generate structures
[2025-03-06 18:52:21,519][gen_pyxtal][INFO] Structure ID 0: (8,) Space group: 31 --> 31 Pmn2_1
[2025-03-06 18:52:21,525][gen_pyxtal][INFO] Structure ID 1: (8,) Space group: 198 --> 198 P2_13
[2025-03-06 18:52:21,554][gen_pyxtal][INFO] Structure ID 2: (8,) Space group: 4 --> 4 P2_1
[2025-03-06 18:52:21,580][gen_pyxtal][INFO] Structure ID 3: (8,) Space group: 193 --> 191 P6/mmm
[2025-03-06 18:52:21,581][gen_pyxtal][WARNING] Compoisition [8] not compatible with symmetry 172: spg = 172 retry.
[2025-03-06 18:52:21,625][gen_pyxtal][INFO] Structure ID 4: (8,) Space group: 64 --> 64 Cmce
[2025-03-06 18:52:22,013][cryspy_init][INFO] Elapsed time for structure generation: 0:00:00.516183
Several output files are also generated.
cryspy.out): Short log. only version 0.10.3 or earlier.cryspy.stat: Status file.data/init_POSCARS: Initial struture file in POSCAR format.
You can open this file using VESTAdata/pkl_data: Directory to save pickled data.log_cryspy: log.err_cryspy: error and warning.Let’s take a look at cryspy.stat file.
[status]
id_queueing = 0 1 2 3 4
Structure ID 0 – 4 are queueing because we just generated structures, and have not submitted yet.
Check the initial structures, if the distance between atoms is too close, you should set the mindist in cryspy.in.
2024年4月21日更新,日本語化
CrySPYはcryspy.statファイルがあれば自動的に計算を再開する.
crypy.statがあれば続きから再開.
はじめから計算をしたければcryspy.statを削除する.
cryspyをもう一度実行する.
cryspy
画面かlog_cryspyファイルに下記のように出力される.
[2023-07-10 18:52:51,859][cryspy_restart][INFO]
Restart CrySPY 1.2.0
[2023-07-10 18:52:51,869][ctrl_job][INFO] # ---------- job status
[2023-07-10 18:52:51,904][ctrl_job][INFO] ID 0: submit job, Stage 1
[2023-07-10 18:52:51,931][ctrl_job][INFO] ID 1: submit job, Stage 1
cryspy.statでステータスが確認できる.
...
(omit)
...
[status]
id_queueing = 2 3 4
id 0 = Stage 1
id 1 = Stage 1
cryspy.inでnjob = 2に設定されているので,CrySPYは構造ID 0と1の二つのジョブをサブミットした.
計算はworkディレクトリの中で行われる.各構造IDのディレクトリが作られる.
tree -d work
work
├── 000000
├── 000001
└── fin
二つのジョブが終了したら,もう一度cryspyを実行する.
cryspy
[2023-07-10 18:55:01,053][cryspy_restart][INFO]
Restart CrySPY 1.2.0
[2023-07-10 18:55:01,058][ctrl_job][INFO] # ---------- job status
[2023-07-10 18:55:01,058][ctrl_job][INFO] ID 0: Stage 1 Done!
[2023-07-10 18:55:01,093][ctrl_job][INFO] collect results: E = -0.00696997755502915 eV/atom
[2023-07-10 18:55:01,132][ctrl_job][INFO] ID 1: Stage 1 Done!
[2023-07-10 18:55:01,133][ctrl_job][INFO] collect results: E = 0.4934076667166454 eV/atom
[2023-07-10 18:55:01,144][cryspy][INFO]
recheck 1
[2023-07-10 18:55:01,145][ctrl_job][INFO] # ---------- job status
[2023-07-10 18:55:01,153][ctrl_job][INFO] ID 2: submit job, Stage 1
[2023-07-10 18:55:01,161][ctrl_job][INFO] ID 3: submit job, Stage 1
もしnstage = 2のようにnstageを2以上に設定していれば,ID 0と1のstage 2のジョブがサブミットされる.
今回はnstage = 1なので,ID 0と1の計算データを収集して,次のIDのジョブをサブミットする.
計算が終わった構造のディレクトリはfinディレクトリに移動される.
5構造全ての計算が終わるまでcryspyを繰り返し実行する.
すべての計算が終わって,計算結果の詳細が必要なければworkディレクトリを削除しても良い.
何度も何度もcryspyを繰り返し実行する時は,自動スクリプト(repeat_cryspy)が役に立つ.
Move to data directory. There should be a few more files.
$ cd data
$ ls
cryspy_rslt cryspy_rslt_energy_asc init_POSCARS opt_POSCARS pkl_data/
cryspy_rslt: Result file.cryspy_rslt_energy_asc: Result file sorted in energy ascending order.init_POSCARS: Initial struture file in POSCAR format.opt_POSCARS: Optimized structure file in POSCAR format.pkl_data/: Directory to save pickled data.The results are written to text files, cryspy_rslt and cryspy_rslt_energy_asc (and also saved in pickle data in pkl_data directory).
Each result appends to cryspy_rslt file in the order in which one finished earlier.
cat cryspy_rslt
Spg_num Spg_sym Spg_num_opt Spg_sym_opt E_eV_atom Magmom Opt
0 139 I4/mmm 139 I4/mmm -3.000850 NaN done
1 98 I4_122 12 C2/m -3.978441 NaN not_yet
2 16 P222 16 P222 -3.348616 NaN not_yet
3 36 Cmc2_1 36 Cmc2_1 -3.520306 NaN not_yet
4 36 Cmc2_1 4 P2_1 -3.304168 NaN not_yet
Not ID order in cryspy_rslt
In cryspy_rslt_energy_asc file, the results are sorted in energy ascending order.
cat cryspy_rslt_energy_asc
Spg_num Spg_sym Spg_num_opt Spg_sym_opt E_eV_atom Magmom Opt
1 98 I4_122 12 C2/m -3.978441 NaN not_yet
3 36 Cmc2_1 36 Cmc2_1 -3.520306 NaN not_yet
2 16 P222 16 P222 -3.348616 NaN not_yet
4 36 Cmc2_1 4 P2_1 -3.304168 NaN not_yet
0 139 I4/mmm 139 I4/mmm -3.000850 NaN done
Spg_num and Spg_sym show space group information on initial structures.
Spg_num_opt and Spg_sym_opt are those of optimized structures.
The last column Opt indicates whether or not optimization reached required accuracy.
Of course only 5 structures are not enough to find stable structures. You can append structures whenever you want. Here let’s append more 5 structures.
For Si-Si mindist, the default value of 1.11 Å is used in the first structure generation (see log_cryspy), which is a little too close.
Let us try to set the mindist to 2.0 Å.
For mindist, see also Features > Restriction on interatomic distances.
Edit cryspy.in and change the value of tot_struc into 10, and add mindist_1 = 2.0
emacs cryspy.in
cat cryspy.in
[basic]
algo = RS
calc_code = soiap
tot_struc = 10
nstage = 1
njob = 2
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Si
nat = 8
mindist_1 = 2.0
[soiap]
soiap_infile = soiap.in
soiap_outfile = soiap.out
soiap_cif = initial.cif
[option]
Then run cryspy, and check log_cryspy file.
cryspy &
cat log_cryspy
...
(omit)
...
2023/03/19 00:01:47
CrySPY 1.0.0
Restart cryspy.py
Changed tot_struc from 5 to 10
Changed mindist from None to [[2.0]]
Backup data
# ---------- Append structures
# ------ mindist
Si - Si 2.0
Structure ID 5 was generated. Space group: 218 --> 221 Pm-3m
Structure ID 6 was generated. Space group: 86 --> 129 P4/nmm
Structure ID 7 was generated. Space group: 129 --> 129 P4/nmm
Structure ID 8 was generated. Space group: 191 --> 191 P6/mmm
Structure ID 9 was generated. Space group: 31 --> 31 Pmn2_1
Remember that CrySPY goes into structure generation mode whenever you change the value of tot_struc.
In this mode, CrySPY does not do any other action such as collecting data, submitting jobs, and so on.
Structure generation mode whenever you change the value of tot_struc.
From version 1.0.0, CrySPY automatically backs up when adding structures.
See features/backup.
Repeat cryspy & several times until all appended structures are done.
The auto script may help you.
ここでは,CrySPYのデータをローカルPCで解析・可視化することを前提としている.
CrySPYをスーパーコンピュータやワークステーションで使用している場合は,データをローカルPCにダウンロードすること.
work や backup ディレクトリは,ファイルサイズが非常に大きくなる可能性があるため,不要であれば削除してよい.
先ほどダウンロードした結果の中にある data/ ディレクトリに移動する.
その後,CrySPY utilityがローカルにダウンロード してある場合は cryspy_analyzer_RS.ipynb をコピーする.
またはGitHubから直接ダウンロードしてくる(CrySPY_utility/notebook/).
Jupyter を起動し(VScode,Jupyter Lab,Jupyter Notebook など),
セルを順番に実行するだけで,以下のような図を得ることができる.

下記のどれか一つに従い,その後CrySPY実行のセクションに移ること.
2025年7月11日 更新
ここで利用しているファイルはCrySPY_utility/examples/ase_Cu8Au8_EAからダウンロードできる. このチュートリアルでは,計算が軽いASEのPure Python EMT calculatorを用いてローカルPCで動作確認を行う.対象となるシステムはCu8Au8.
cryspy.inの例.
[basic]
algo = EA
calc_code = ASE
nstage = 1
njob = 5
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Cu Au
nat = 8 8
[EA]
n_pop = 10
n_crsov = 5
n_perm = 2
n_strain = 2
n_rand = 1
n_elite = 1
n_fittest = 5
slct_func = TNM
t_size = 2
maxgen_ea = 0
[ASE]
ase_python = ase_in.py
[option]
algo = EA を使用.jobcmdをbashに変更.tot_strucは使用しない.構造数はn_popで決まる.n_pop = n_crsov + n_perm + n_strain + n_randcalc_in/以下はチュートリアル > ランダムサーチ(RS) > ASE in your local PCと同様
from ase.constraints import FixSymmetry
from ase.filters import FrechetCellFilter
from ase.calculators.emt import EMT
from ase.optimize import BFGS
from ase.io import read, write
# ---------- input structure
# CrySPY outputs 'POSCAR' as an input file in work/xxxxxx directory
atoms = read('POSCAR', format='vasp')
# ---------- setting and run
atoms.calc = EMT()
atoms.set_constraint([FixSymmetry(atoms)])
cell_filter = FrechetCellFilter(atoms, hydrostatic_strain=False)
opt = BFGS(cell_filter)
# ---------- run
converged = opt.run(fmax=0.01, steps=2000)
# ---------- rule in ASE interface
# output file for energy: 'log.tote' in eV/cell
# CrySPY reads the last line of 'log.tote' file
# outimized structure: 'CONTCAR' file in vasp format
# check_opt: 'out_check_opt' file ('done' or 'not yet')
# CrySPY reads the last line of 'out_check_opt' file
# ------ energy
e = cell_filter.atoms.get_total_energy() # eV/cell
with open('log.tote', mode='w') as f:
f.write(str(e))
# ------ struc
opt_atoms = cell_filter.atoms.copy()
opt_atoms.set_constraint(None) # remove constraint for pymatgen
write('CONTCAR', opt_atoms, format='vasp', direct=True)
# ------ check_opt
with open('out_check_opt', mode='w') as f:
if converged:
f.write('done\n')
else:
f.write('not yet\n')
#!/bin/sh
# ---------- ASE
python3 ase_in.py > out.log
2025年4月6日
cryspyを実行すると,構造生成モードになり,第一世代のランダム構造が生成されて,一旦プログラムは終了する.
cryspy
出力
...
[2025-04-06 09:15:34,720][ea_init][INFO] # ---------- Initialize evolutionary algorithm
[2025-04-06 09:15:34,720][ea_init][INFO] # ------ Generation 1
[2025-04-06 09:15:34,720][ea_init][INFO] 10 structures by random
cryspy.statを見ると,EAでは現在の世代の情報が追加されている.
[status]
generation = 1
id_queueing = 0 1 2 3 4 5 6 7 8 9
何度かcryspyを実行して,第一世代の構造最適化を終わらせると,下記のように出力される.
...
[2025-04-06 09:20:26,218][ctrl_job][INFO] Done generation 1
[2025-04-06 09:20:26,218][ctrl_job][INFO]
EA is ready
準備ができた状態でもう一度cryspyを実行すると,バックアップが実行されてから,次世代構造の生成が始まる.
cryspy
...
[2025-04-06 09:35:11,546][cryspy_restart][INFO] read input, cryspy.in
[2025-04-06 09:35:11,554][ctrl_job][INFO] # ---------- job status
[2025-04-06 09:35:11,554][ctrl_job][INFO] Done generation 1
[2025-04-06 09:35:11,554][utility][INFO] Backup data
[2025-04-06 09:35:11,611][ea_next_gen][INFO] # ---------- Evolutionary algorithm
[2025-04-06 09:35:11,611][ea_next_gen][INFO] Generation 2
[2025-04-06 09:35:11,613][ea_next_gen][INFO] # ------ natural selection
[2025-04-06 09:35:11,687][ea_next_gen][INFO] ranking without duplication (including elite):
[2025-04-06 09:35:11,687][ea_next_gen][INFO] Structure ID 1, fitness: -0.00530
[2025-04-06 09:35:11,687][ea_next_gen][INFO] Structure ID 3, fitness: 0.01490
[2025-04-06 09:35:11,687][ea_next_gen][INFO] Structure ID 4, fitness: 0.04485
[2025-04-06 09:35:11,687][ea_next_gen][INFO] Structure ID 7, fitness: 0.11501
[2025-04-06 09:35:11,687][ea_next_gen][INFO] Structure ID 8, fitness: 0.15254
[2025-04-06 09:35:11,687][ea_next_gen][INFO] # ------ Generate children
[2025-04-06 09:35:11,687][ea_child][INFO] # -- mindist
[2025-04-06 09:35:11,689][struc_util][INFO] Cu - Cu: 1.32
[2025-04-06 09:35:11,689][struc_util][INFO] Cu - Au: 1.34
[2025-04-06 09:35:11,689][struc_util][INFO] Au - Au: 1.36
[2025-04-06 09:35:11,740][crossover][INFO] Structure ID 10 (8, 8) was generated from 3 and 1 by crossover. Space group: 1 P1
[2025-04-06 09:35:11,764][crossover][WARNING] remove_within_mindist: some atoms within mindist. retry.
[2025-04-06 09:35:11,774][crossover][INFO] Structure ID 11 (8, 8) was generated from 3 and 1 by crossover. Space group: 1 P1
[2025-04-06 09:35:11,789][crossover][INFO] Structure ID 12 (8, 8) was generated from 1 and 4 by crossover. Space group: 1 P1
[2025-04-06 09:35:11,833][crossover][INFO] Structure ID 13 (8, 8) was generated from 1 and 3 by crossover. Space group: 1 P1
[2025-04-06 09:35:11,852][crossover][WARNING] mindist in _add_border_line: Cu - Cu, 0.567032320824818. retry.
[2025-04-06 09:35:11,861][crossover][INFO] Structure ID 14 (8, 8) was generated from 7 and 1 by crossover. Space group: 1 P1
[2025-04-06 09:35:11,875][permutation][INFO] Structure ID 15 (8, 8) was generated from 1 by permutation. Space group: 146 R3
[2025-04-06 09:35:11,888][permutation][INFO] Structure ID 16 (8, 8) was generated from 3 by permutation. Space group: 1 P1
[2025-04-06 09:35:11,890][strain][WARNING] mindist in strain: Cu - Cu, 1.3050485787603692. retry.
[2025-04-06 09:35:11,903][strain][INFO] Structure ID 17 (8, 8) was generated from 3 by strain. Space group: 1 P1
[2025-04-06 09:35:11,917][strain][INFO] Structure ID 18 (8, 8) was generated from 1 by strain. Space group: 1 P1
[2025-04-06 09:35:12,513][ea_child][INFO] # ------ Random structure generation
[2025-04-06 09:35:12,513][rs_gen][INFO] # ------ mindist
[2025-04-06 09:35:12,515][struc_util][INFO] Cu - Cu: 1.32
[2025-04-06 09:35:12,515][struc_util][INFO] Cu - Au: 1.34
[2025-04-06 09:35:12,515][struc_util][INFO] Au - Au: 1.36
[2025-04-06 09:35:12,516][rs_gen][INFO] # ------ generate structures
[2025-04-06 09:35:12,530][gen_pyxtal][INFO] Structure ID 19: (8, 8) Space group: 86 --> 86 P4_2/n
[2025-04-06 09:35:12,533][ea_next_gen][INFO] # ------ Select elites
[2025-04-06 09:35:12,533][ea_next_gen][INFO] Structure ID 9 keeps as the elite
あとはcryspyの実行を繰り返せば,探索が進む.
以下は第3世代まで計算を終えたcryspy_rsltの例である.EAでは世代の情報(Gen)も追加される.
Gen Spg_num Spg_sym Spg_num_opt Spg_sym_opt E_eV_atom Magmom Opt
0 1 214 I4_132 230 Ia-3d 1.168743 NaN no_file
1 1 198 P2_13 198 P2_13 -0.005303 NaN no_file
2 1 95 P4_322 95 P4_322 0.389566 NaN no_file
3 1 27 Pcc2 27 Pcc2 0.014898 NaN no_file
4 1 60 Pbcn 60 Pbcn 0.044852 NaN no_file
5 1 116 P-4c2 116 P-4c2 0.403246 NaN no_file
6 1 187 P-6m2 187 P-6m2 1.054706 NaN no_file
7 1 161 R3c 160 R3m 0.115009 NaN no_file
8 1 146 R3 146 R3 0.152535 NaN no_file
9 1 60 Pbcn 47 Pmmm -0.005676 NaN no_file
10 2 1 P1 1 P1 0.026070 NaN no_file
11 2 1 P1 7 Pc 0.005898 NaN no_file
12 2 1 P1 1 P1 0.005208 NaN no_file
13 2 1 P1 1 P1 0.005506 NaN no_file
14 2 1 P1 1 P1 0.024364 NaN no_file
15 2 146 R3 146 R3 0.011525 NaN no_file
16 2 1 P1 1 P1 0.014590 NaN no_file
17 2 1 P1 1 P1 0.015236 NaN no_file
18 2 1 P1 2 P-1 -0.012335 NaN no_file
19 2 86 P4_2/n 140 I4/mcm 0.274548 NaN no_file
20 3 1 P1 1 P1 0.013611 NaN no_file
21 3 1 P1 10 P2/m -0.014166 NaN no_file
22 3 1 P1 1 P1 0.019472 NaN no_file
23 3 1 P1 1 P1 0.011641 NaN no_file
24 3 1 P1 1 P1 0.000297 NaN no_file
25 3 1 P1 1 P1 0.005596 NaN no_file
26 3 1 P1 1 P1 0.013374 NaN no_file
27 3 1 P1 2 P-1 0.005055 NaN no_file
28 3 2 P-1 12 C2/m -0.012396 NaN no_file
29 3 182 P6_322 182 P6_322 0.711472 NaN no_file
各世代ごとに用いたEAのパラメーターがea_infoに出力される.
Gen Population Crossover Permutation Strain Random Elite crs_lat slct_func
1 10 0 0 0 10 0 random TNM
2 10 5 2 2 1 1 random TNM
3 10 5 2 2 1 1 random TNM
構造生成手法や親個体の情報が,ea_originに出力される.
Gen Struc_ID Operation Parent
1 0 random None
1 1 random None
1 2 random None
1 3 random None
1 4 random None
1 5 random None
1 6 random None
1 7 random None
1 8 random None
1 9 random None
2 10 crossover (3, 1)
2 11 crossover (3, 1)
2 12 crossover (1, 4)
2 13 crossover (1, 3)
2 14 crossover (7, 1)
2 15 permutation (1,)
2 16 permutation (3,)
2 17 strain (3,)
2 18 strain (1,)
2 19 random None
2 9 elite elite
3 20 crossover (18, 12)
3 21 crossover (12, 9)
3 22 crossover (12, 18)
3 23 crossover (18, 9)
3 24 crossover (13, 18)
3 25 permutation (18,)
3 26 permutation (9,)
3 27 strain (18,)
3 28 strain (18,)
3 29 random None
3 18 elite elite
ここでは,CrySPYのデータをローカルPCで解析・可視化することを前提としている.
CrySPYをスーパーコンピュータやワークステーションで使用している場合は,データをローカルPCにダウンロードすること.
work や backup ディレクトリは,ファイルサイズが非常に大きくなる可能性があるため,不要であれば削除してよい.
先ほどダウンロードした結果の中にある data/ ディレクトリに移動する.
その後,CrySPY utilityがローカルにダウンロード してある場合は cryspy_analyzer_EA.ipynb をコピーする.
またはGitHubから直接ダウンロードしてくる(CrySPY_utility/notebook/).
Jupyter を起動し(VScode,Jupyter Lab,Jupyter Notebook など),
セルを順番に実行するだけで,以下のような図を得ることができる.

動作環境
下記のどれか一つに従い,その後CrySPY実行のセクションに移ること.
2025年7月11日 更新
ここで利用しているファイルはCrySPY_utility/examples/ase_Cu-Ag-Au_EA-vcからダウンロードできる. このチュートリアルでは,計算が軽いASEのPure Python EMT calculatorを用いてローカルPCで動作確認を行う.対象となるシステムは3元系のCu-Ag-Au.
cryspy.inの例.
[basic]
algo = EA-vc
calc_code = ASE
nstage = 1
njob = 5
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Cu Ag Au
ll_nat = 0 0 0
ul_nat = 8 8 8
[ASE]
ase_python = ase_in.py
[EA]
n_pop = 20
n_crsov = 5
n_perm = 2
n_strain = 2
n_rand = 2
n_add = 3
n_elim = 3
n_subs = 3
target = random
n_elite = 2
n_fittest = 10
slct_func = TNM
t_size = 2
end_point = 0.0 0.0 0.0
maxgen_ea = 0
[option]
algo = EA-vc を使用.jobcmdをbashに変更.ll_natで各元素の原子数の最小値,ul_natで最大値を指定.n_pop = n_crsov + n_perm + n_strain + n_rand + n_add + n_elim + n_subsend_pointにはCu, Ag, Auにおける各単体の1原子当たりのエネルギー(eV/atom)を入力.ASEのPure Python EMT calculatorは単体のエネルギーが0になるので今回は0.0.(CHGNetを使う場合の例はASE-CHGNet(Cu-Au))calc_in/以下はチュートリアル > ランダムサーチ(RS) > ASE in your local PCと同様
from ase.constraints import FixSymmetry
from ase.filters import FrechetCellFilter
from ase.calculators.emt import EMT
from ase.optimize import BFGS
import numpy as np
from ase.io import read, write
# ---------- input structure
# CrySPY outputs 'POSCAR' as an input file in work/xxxxxx directory
atoms = read('POSCAR', format='vasp')
# ---------- setting and run
atoms.calc = EMT()
atoms.set_constraint([FixSymmetry(atoms)])
cell_filter = FrechetCellFilter(atoms, hydrostatic_strain=False)
opt = BFGS(cell_filter)
# ---------- run
converged = opt.run(fmax=0.01, steps=2000)
# ---------- rule in ASE interface
# output file for energy: 'log.tote' in eV/cell
# CrySPY reads the last line of 'log.tote' file
# outimized structure: 'CONTCAR' file in vasp format
# check_opt: 'out_check_opt' file ('done' or 'not yet')
# CrySPY reads the last line of 'out_check_opt' file
# ------ energy
e = cell_filter.atoms.get_total_energy() # eV/cell
with open('log.tote', mode='w') as f:
f.write(str(e))
# ------ struc
opt_atoms = cell_filter.atoms.copy()
opt_atoms.set_constraint(None) # remove constraint for pymatgen
write('CONTCAR', opt_atoms, format='vasp', direct=True)
# ------ check_opt
with open('out_check_opt', mode='w') as f:
if converged:
f.write('done\n')
else:
f.write('not yet\n')
#!/bin/sh
# ---------- ASE
python3 ase_in.py > out.log
2025年7月11日 更新
CHGNetのインストールが必要
ここで利用しているファイルはCrySPY_utility/examples/ase_chgnet_Cu-Au_EA-vcからダウンロードできる. このチュートリアルでは,機械学習ポテンシャルのCHGNetを用いてジョブスケジューラーを備えた計算機クラスタで行うことを想定している. 手持ちのPCでも十分動作するので,そうしたい場合は入力を適宜変更すること. 対象となるシステムは2元系のCu-Au.
EA-vcではcryspy.inでend_pointとして,各元素単体の1原子当たりのエネルギーを基準として用いるため,事前に計算する必要がある.
exampleファイルの中に二つのディレクトリがあるはず.
Au_fcc
├── POSCAR
├── chgnet_in.py
└── job_cryspy
Cu_fcc
├── POSCAR
├── chgnet_in.py
└── job_cryspy
結晶構造データ(POSCAR)とその構造最適化を行いエネルギーを求めるpythonスクリプト(chgnet_in.py)およびジョブスクリプト(job_cryspy)があるので適宜使用する計算機の環境に合わせて編集すること.
ジョブを実行する(ジョブサブミットのコマンドは適宜読み替えること).
cd Au_fcc
qsub job_cryspy
cd ../Cu_fcc
qsub job_cryspy
cd ..
計算が無事に終わったら,それぞれのディレクトリにend_pointというファイルができており,ここに構造最適化後の1原子あたりのエネルギー(eV/atom)が出力されている.
cat Au_fcc/end_point
-3.2357187271118164
cat Cu_fcc/end_point
-4.083529472351074
これらをcryspy.inでの入力に用いる.
cryspy.inの例.
[basic]
algo = EA-vc
calc_code = ASE
nstage = 1
njob = 20
jobcmd = qsub
jobfile = job_cryspy
[structure]
atype = Cu Au
ll_nat = 0 0
ul_nat = 8 8
[ASE]
ase_python = chgnet_in.py
[EA]
n_pop = 20
n_crsov = 5
n_perm = 2
n_strain = 2
n_rand = 2
n_add = 3
n_elim = 3
n_subs = 3
target = random
n_elite = 2
n_fittest = 10
slct_func = TNM
t_size = 2
maxgen_ea = 0
end_point = -4.08352709 -3.23571777
[option]
algo = EA-vc を使用.jobcmdは計算機環境に合わせて変更すること.ll_natで各元素の原子数の最小値,ul_natで最大値を指定.n_pop = n_crsov + n_perm + n_strain + n_rand + n_add + n_elim + n_subsend_pointには事前計算しておいたCu, Auにおける各単体の1原子当たりのエネルギー(eV/atom)を入力.atypeの順番.calc_in/以下はチュートリアル > ランダムサーチ(RS) > ASE in your local PCと同様であるが,少しだけCHGNet用に変更している. ジョブスクリプトのpython本体のpathも計算機環境に合わせること.
# ---------- import
from ase.constraints import FixSymmetry
from ase.filters import FrechetCellFilter
from ase.io import read, write
from ase.optimize import FIRE, BFGS, LBFGS
from chgnet.model import CHGNetCalculator
# ---------- input structure
# CrySPY outputs 'POSCAR' as an input file in work/xxxxxx directory
atoms = read('POSCAR')
# ---------- set up
atoms.calc = CHGNetCalculator()
atoms.set_constraint([FixSymmetry(atoms)])
cell_filter = FrechetCellFilter(atoms)
opt = BFGS(cell_filter, trajectory='opt.traj')
# ---------- run
converged = opt.run(fmax=0.01, steps=2000)
# ---------- rule in ASE interface
# output file for energy: 'log.tote' in eV/cell
# CrySPY reads the last line of 'log.tote' file
# outimized structure: 'CONTCAR' file in vasp format
# check_opt: 'out_check_opt' file ('done' or 'not yet')
# CrySPY reads the last line of 'out_check_opt' file
# ------ energy
e = cell_filter.atoms.get_total_energy() # eV/cell
with open('log.tote', mode='w') as f:
f.write(str(e))
# ------ struc
opt_atoms = cell_filter.atoms.copy()
opt_atoms.set_constraint(None) # remove constraint for pymatgen
write('CONTCAR', opt_atoms, format='vasp', direct=True)
# ------ check_opt
with open('out_check_opt', mode='w') as f:
if converged:
f.write('done\n')
else:
f.write('not yet\n')
#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
####$ -V -S /bin/zsh
#$ -N CuAu_CrySPY_ID
#$ -pe smp 2
# ---------- OpenMP
export OMP_NUM_THREADS=2
# ---------- ASE
/usr/local/Python-3.10.13/bin/python3 chgnet_in.py > out.log
2025年7月12日
バージョン1.4.2でEA-vcはVASPに対応.
ここで利用しているファイルはCrySPY_utility/examples/vasp_Fe-Al_EA-vcからダウンロードできる. このチュートリアルでは,VASPを用いてジョブスケジューラーを備えた計算機クラスタで行うことを想定している. 対象となるシステムは2元系のFe-Alで,強磁性を仮定した計算を行う.
EA-vcではcryspy.inでend_pointとして,各元素単体の1原子当たりのエネルギーを基準として用いるため,事前に計算する必要がある.
exampleファイルの中に二つのディレクトリがあるはず.
Al-fcc
├── POSCAR
├── INCAR
├── POTCAR_dummy
└── job_cryspy
Fe-bcc
├── POSCAR
├── INCAR
├── POTCAR_dummy
└── job_cryspy
結晶構造データ(POSCAR)とその構造最適化を行いエネルギーを求めるインプットファイル(INCAR)およびジョブスクリプト(job_cryspy)があるので適宜使用する計算機の環境に合わせて編集すること.
また,POTCARファイルは配布できないので各自で準備すること.
INCARファイルでは,最終的に使用するカットオフなどの値を単体の計算でも利用しなければならないので注意すること.
ジョブを実行する(ジョブサブミットのコマンドは適宜読み替えること).
cd Al_fcc
qsub job_cryspy
cd ../Fe_bcc
qsub job_cryspy
cd ..
計算が終わったら1原子あたりのエネルギーを計算しておく. 今回はBCCやFCCなので,ユニットセルは1原子で構成されており,全エネルギーの値をそのまま使えばよい. 通常はユニットセルの原子数でセルあたりのエネルギーを割って計算する.
cryspy.inの例.
[basic]
algo = EA-vc
calc_code = VASP
nstage = 2
njob = 10
jobcmd = qsub
jobfile = job_cryspy
[structure]
atype = Fe Al
ll_nat = 0 0
ul_nat = 8 8
[VASP]
kppvol = 40 120
vasp_MAGMOM = 4.0 0.0
[EA]
n_pop = 20
n_crsov = 5
n_perm = 2
n_strain = 2
n_rand = 2
n_add = 3
n_elim = 3
n_subs = 3
target = random
n_elite = 2
n_fittest = 10
slct_func = TNM
t_size = 2
maxgen_ea = 0
end_point = -8.24249611 -3.74226843
[option]
algo = EA-vc を使用.jobcmdは計算機環境に合わせて変更すること.ll_natで各元素の原子数の最小値,ul_natで最大値を指定.n_pop = n_crsov + n_perm + n_strain + n_rand + n_add + n_elim + n_subsend_pointには事前計算しておいた,FeおよびAlにおける各単体の1原子当たりのエネルギー(eV/atom)を入力.atypeの順番.[VASP]セクションのvasp_MAGMOMにはatypeに対応する元素のMAGMOMの値を入れる.
CrySPYは./work/xxx/INCAR(xxxは構造ID)にINCARをコピーする際に,n*vasp_MAGMOM(nは原子数)を追記する.
例えば, atype = (‘Fe’, ‘Al’),nat = (5, 3),vasp_MAGMOM = (4.0 0.0)の場合,下記のように追記する.
MAGMOM = 5*4.0 3*0.0
このチュートリアルでは使用していないが,他にもINCARのLDAUL,LDAUUおよびLDAUJに対応している.cryspy.inではvasp_をつけて下記のように使う.
[VASP]
kppvol = 40 120
vasp_MAGMOM = 4.0 0.0
vasp_LDAUL = 2 -1
vasp_LDAUU = 4.0 0.0
vasp_LDAUJ = 0.0 0.0
nat = (5, 0)のように2元系から単体に次元が落ちる場合,CrySPYは./work/xxx/INCARに下記のように追記する.
原子数がゼロの元素に関しては何もしない.
MAGMOM = 5*4.0
LDAUL = 2
LDAUU = 4.0
LDAUJ = 0.0
EA-vcでは各元素に対応するPOTCARファイルをそれぞれ準備する. ファイル名は
POTCAR_FePOTCAR_Alのように末尾にatypeに書いた元素名をつける.
CrySPYは計算する構造の原子数に合わせて,./work/xxx/POTCARを準備する.
つまり,nat = (5, 0)のようにFeだけから構成される構造の場合はPOTCAR_Fe,
nat = (4, 4)のようにFe-Al系の場合はPOTCAR_FeとPOTCAR_Alを繋げてPOTCARにする.
#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
####$ -V -S /bin/zsh
#$ -N FeAl_CrySPY_ID
#$ -pe smp 32
# ---------- vasp
VASPROOT=/usr/local/vasp/vasp.6.4.2/bin
mpirun -np $NSLOTS $VASPROOT/vasp_std
1_INCAR
SYSTEM = FeAl
Algo = Fast
####LREAL = Auto
ENCUT = 348
ISMEAR = 1
SIGMA = 0.1
NSW = 40
IBRION = 2
ISIF = 2
ISPIN = 2
######MAGMOM = # cryspy append MAGMOM in work/xx/INCAR
EDIFF = 1e-6
EDIFFG = -0.01
KPAR = 4
LWAVE = .FALSE.
LCHARG = .FALSE.
2_INCAR
SYSTEM = FeAl
Algo = Fast
####LREAL = Auto
ENCUT = 348
ISMEAR = 1
SIGMA = 0.1
NSW = 200
IBRION = 2
ISIF = 3
ISPIN = 2
######MAGMOM = # cryspy append MAGMOM in work/xx/INCAR
EDIFF = 1e-6
EDIFFG = -0.01
KPAR = 4
LWAVE = .FALSE.
LCHARG = .FALSE.
2025年7月18日
バージョン1.4.2でEA-vcはQEに対応.
ここで利用しているファイルはCrySPY_utility/examples/qe_Fe-Al_EA-vcからダウンロードできる. このチュートリアルでは,QEを用いてジョブスケジューラーを備えた計算機クラスタで行うことを想定している. 対象となるシステムは2元系のFe-Alで,強磁性を仮定した計算を行う.
EA-vcではcryspy.inでend_pointとして,各元素単体の1原子当たりのエネルギーを基準として用いるため,事前に計算する必要がある.
exampleファイルの中に二つのディレクトリがあるはず.
Al-fcc
├── pwscf.in
└── job_cryspy
Fe-bcc
├── pwscf.in
└── job_cryspy
インプットファイル(pwscf.in)およびジョブスクリプト(job_cryspy)があるので適宜使用する計算機の環境に合わせて編集すること.
pwscf.inファイルでは,最終的に使用するカットオフなどの値を単体の計算でも利用しなければならないので注意すること.
ジョブを実行する(ジョブサブミットのコマンドは適宜読み替えること).
cd Al_fcc
qsub job_cryspy
cd ../Fe_bcc
qsub job_cryspy
cd ..
計算が終わったら1原子あたりのエネルギーをeV単位に変換しておくこと. 今回はBCCやFCCなので,ユニットセルは1原子で構成されているが, 通常はユニットセルの原子数でセルあたりのエネルギーを割って計算する.
cryspy.inの例.
[basic]
algo = EA-vc
calc_code = QE
nstage = 2
njob = 10
jobcmd = qsub
jobfile = job_cryspy
[structure]
atype = Fe Al
ll_nat = 0 0
ul_nat = 8 8
[QE]
kppvol = 40 120
qe_infile = pwscf.in
qe_outfile = pwscf.out
[EA]
n_pop = 20
n_crsov = 5
n_perm = 2
n_strain = 2
n_rand = 2
n_add = 3
n_elim = 3
n_subs = 3
target = random
n_elite = 2
n_fittest = 10
slct_func = TNM
t_size = 2
maxgen_ea = 6
end_point = -3406.14117375 -91.25463006
[option]
algo = EA-vc を使用.jobcmdは計算機環境に合わせて変更すること.ll_natで各元素の原子数の最小値,ul_natで最大値を指定.n_pop = n_crsov + n_perm + n_strain + n_rand + n_add + n_elim + n_subsend_pointには事前計算しておいた,FeおよびAlにおける各単体の1原子当たりのエネルギー(eV/atom)を入力.atypeの順番.VASPと違ってQEでは組成可変型への対応は簡単で,2元系の探索の場合は常に2元系のインプット,3元系なら常に3元系のインプットを./calc_inに準備すればよい.
ただしnatだけは変更の必要があるので,CrySPYが自動で書き換える.
準備する際は下記のように何か適当な数字でnatを書いておく.
nat = 1
CrySPYは./work/xxx/において,先頭の空白を除いてnatから始まる行を
nat = (実際の原子数)
に自動的に書き換える.
#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
####$ -V -S /bin/zsh
#$ -N FeAl_CrySPY_ID
#$ -pe smp 32
QEROOT=/usr/local/qe/q-e-qe-7.3.1/bin
mpirun -np $NSLOTS $QEROOT/pw.x -nk 4 < pwscf.in > pwscf.out
if [ -e "CRASH" ]; then
sed -i -e '3 s/^.*$/skip/' stat_job
exit 1
fi
1_pwscf.in
&control
calculation = 'relax'
nstep = 40
pseudo_dir = '/usr/local/qe/gbrv/all_pbe_UPF_v1.5/'
outdir='./outdir/'
/
&system
ibrav = 0
nat = 1
ntyp = 2
ecutwfc = 40
ecutrho = 200
occupations = "smearing"
smearing = "mp"
degauss = 0.01
nspin = 2
starting_magnetization(1) = 0.4
starting_magnetization(2) = 0.0
/
&electrons
mixing_beta = 0.4
/
&ions
/
&cell
/
ATOMIC_SPECIES
Fe -1.0 fe_pbe_v1.5.uspp.F.UPF
Al -1.0 al_pbe_v1.uspp.F.UPF
2_pwscf.in
&control
calculation = 'vc-relax'
nstep = 200
pseudo_dir = '/usr/local/qe/gbrv/all_pbe_UPF_v1.5/'
outdir='./outdir/'
/
&system
ibrav = 0
nat = 1
ntyp = 2
ecutwfc = 40
ecutrho = 200
occupations = "smearing"
smearing = "mp"
degauss = 0.01
nspin = 2
starting_magnetization(1) = 0.4
starting_magnetization(2) = 0.0
/
&electrons
mixing_beta = 0.4
/
&ions
/
&cell
/
ATOMIC_SPECIES
Fe -1.0 fe_pbe_v1.5.uspp.F.UPF
Al -1.0 al_pbe_v1.uspp.F.UPF
2025年6月16日
cryspyを実行すると,構造生成モードになり,第一世代のランダム構造が生成されて,一旦プログラムは終了する.
cryspy
出力をみると,ll_natとul_natの範囲の原子数で構造が生成されているのが確認できる.
...
[2025-06-16 10:04:45,648][cryspy_init][INFO] # ---------- Initial structure generation
[2025-06-16 10:04:45,648][rs_gen][INFO] # ------ mindist
[2025-06-16 10:04:45,650][struc_util][INFO] Cu - Cu: 1.32
[2025-06-16 10:04:45,650][struc_util][INFO] Cu - Ag: 1.385
[2025-06-16 10:04:45,650][struc_util][INFO] Cu - Au: 1.34
[2025-06-16 10:04:45,650][struc_util][INFO] Ag - Ag: 1.45
[2025-06-16 10:04:45,650][struc_util][INFO] Ag - Au: 1.405
[2025-06-16 10:04:45,650][struc_util][INFO] Au - Au: 1.36
[2025-06-16 10:04:45,650][rs_gen][INFO] # ------ generate structures
[2025-06-16 10:04:45,659][gen_pyxtal][WARNING] Compoisition [1 4] not compatible with symmetry 34: spg = 34 retry.
[2025-06-16 10:04:45,662][gen_pyxtal][WARNING] Compoisition [ 2 2 12] not compatible with symmetry 39: spg = 39 retry.
[2025-06-16 10:04:45,691][gen_pyxtal][INFO] Structure ID 0: (3, 1, 2) Space group: 82 --> 119 I-4m2
[2025-06-16 10:04:45,694][gen_pyxtal][WARNING] Compoisition [6 6 2] not compatible with symmetry 57: spg = 57 retry.
[2025-06-16 10:04:45,749][gen_pyxtal][INFO] Structure ID 1: (1, 8, 5) Space group: 71 --> 71 Immm
[2025-06-16 10:04:45,857][gen_pyxtal][INFO] Structure ID 2: (3, 7, 8) Space group: 174 --> 174 P-6
...
cryspy.statを見ると,EAでは現在の世代の情報が追加されている.
[status]
generation = 1
id_queueing = 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19
何度かcryspyを実行して,第一世代の構造最適化を終わらせると,下記のように出力される.
...
[2025-06-16 10:25:56,962][ctrl_job][INFO] Done generation 1
[2025-06-16 10:25:56,962][ctrl_job][INFO] Calculate convex hull for generation 1
[2025-06-16 10:25:57,854][ctrl_job][INFO]
EA is ready
この時点で,./data/convex_hull/にhull distanceの情報とconvex hullのグラフが出力されている.
ID hull distance (eV/atom) Num_atom
7 0.000000 (0, 2, 6)
14 0.036510 (1, 7, 6)
17 0.064702 (0, 1, 5)
19 0.113649 (0, 0, 8)
16 0.168530 (6, 4, 8)
9 0.186497 (8, 4, 6)
1 0.187379 (1, 8, 5)
11 0.233893 (4, 5, 4)
3 0.273365 (6, 5, 5)
10 0.326759 (1, 4, 4)
2 0.330749 (3, 7, 8)
8 0.359543 (6, 2, 7)
4 0.404169 (4, 4, 2)
18 0.422989 (0, 6, 8)
13 0.428456 (0, 6, 3)
5 0.444792 (7, 4, 7)
6 0.464305 (7, 7, 7)
12 0.556654 (3, 0, 0)
15 0.560062 (6, 7, 1)
0 0.644278 (3, 1, 2)

準備ができた状態でもう一度cryspyを実行すると,バックアップが実行されてから,次世代構造の生成が始まる.
cryspy
...
[2025-06-16 10:37:19,860][ctrl_job][INFO] Done generation 1
[2025-06-16 10:37:20,136][utility][INFO] Backup data
[2025-06-16 10:37:20,173][ea_next_gen][INFO] # ---------- Evolutionary algorithm
[2025-06-16 10:37:20,174][ea_next_gen][INFO] Generation 2
[2025-06-16 10:37:20,174][ea_next_gen][INFO] # ------ natural selection
[2025-06-16 10:37:20,177][ea_next_gen][INFO] ranking without duplication (including elite):
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 7, fitness: 0.00000
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 14, fitness: 0.03651
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 17, fitness: 0.06470
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 19, fitness: 0.11365
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 16, fitness: 0.16853
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 9, fitness: 0.18650
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 1, fitness: 0.18738
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 11, fitness: 0.23389
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 3, fitness: 0.27336
[2025-06-16 10:37:20,177][ea_next_gen][INFO] Structure ID 10, fitness: 0.32676
[2025-06-16 10:37:20,177][ea_next_gen][INFO] # ------ Generate children
[2025-06-16 10:37:20,177][ea_child][INFO] # -- mindist
[2025-06-16 10:37:20,179][struc_util][INFO] Cu - Cu: 1.32
[2025-06-16 10:37:20,179][struc_util][INFO] Cu - Ag: 1.385
[2025-06-16 10:37:20,179][struc_util][INFO] Cu - Au: 1.34
[2025-06-16 10:37:20,179][struc_util][INFO] Ag - Ag: 1.45
[2025-06-16 10:37:20,179][struc_util][INFO] Ag - Au: 1.405
[2025-06-16 10:37:20,179][struc_util][INFO] Au - Au: 1.36
[2025-06-16 10:37:20,217][crossover][INFO] Structure ID 20 (0, 4, 7) was generated from 19 and 14 by crossover. Space group: 1 P1
[2025-06-16 10:37:20,219][crossover][INFO] Structure ID 21 (0, 1, 7) was generated from 7 and 17 by crossover. Space group: 1 P1
[2025-06-16 10:37:20,221][crossover][INFO] Structure ID 22 (3, 0, 8) was generated from 16 and 19 by crossover. Space group: 1 P1
[2025-06-16 10:37:20,225][crossover][INFO] Structure ID 23 (0, 1, 7) was generated from 7 and 17 by crossover. Space group: 1 P1
...
[2025-06-16 10:37:20,809][ea_next_gen][INFO] # ------ Select elites
[2025-06-16 10:37:20,809][ea_next_gen][INFO] Structure ID 7 keeps as the elite
[2025-06-16 10:37:20,809][ea_next_gen][INFO] Structure ID 14 keeps as the elite
あとはcryspyの実行を繰り返せば,探索が進む.
ここでは,EAと異なるところを中心に述べる.
以下は第3世代まで計算を終えたcryspy_rsltの例である.EA-vcでは,形成エネルギー(Ef_eV_atom)と原子数(Num_atom)も追加される.
Gen Spg_num Spg_sym Spg_num_opt Spg_sym_opt E_eV_atom Ef_eV_atom Num_atom Magmom Opt
0 1 119 I-4m2 119 I-4m2 0.639865 0.639865 (3, 1, 2) NaN no_file
1 1 71 Immm 71 Immm 0.182650 0.182650 (1, 8, 5) NaN no_file
2 1 174 P-6 187 P-6m2 0.324864 0.324864 (3, 7, 8) NaN no_file
3 1 71 Immm 71 Immm 0.269227 0.269227 (6, 5, 5) NaN no_file
4 1 12 C2/m 65 Cmmm 0.401521 0.401521 (4, 4, 2) NaN no_file
7 1 123 P4/mmm 123 P4/mmm -0.009930 -0.009930 (0, 2, 6) NaN no_file
10 1 107 I4mm 107 I4mm 0.320875 0.320875 (1, 4, 4) NaN no_file
5 1 121 I-42m 121 I-42m 0.439643 0.439643 (7, 4, 7) NaN no_file
6 1 115 P-4m2 115 P-4m2 0.459892 0.459892 (7, 7, 7) NaN no_file
8 1 81 P-4 81 P-4 0.354247 0.354247 (6, 2, 7) NaN no_file
9 1 11 P2_1/m 11 P2_1/m 0.182084 0.182084 (8, 4, 6) NaN no_file
11 1 10 P2/m 10 P2/m 0.229819 0.229819 (4, 5, 4) NaN no_file
原子数の情報はnat_dataにも出力される.
ID ('Cu', 'Ag', 'Au')
0 (3, 1, 2)
1 (1, 8, 5)
2 (3, 7, 8)
3 (6, 5, 5)
4 (4, 4, 2)
5 (7, 4, 7)
6 (7, 7, 7)
7 (0, 2, 6)
8 (6, 2, 7)
9 (8, 4, 6)
10 (1, 4, 4)
...
例えば第3世代終了時点のhull distanceのデータは./convex_hull/hull_dist_all_gen_3というファイルに出力される.
ID hull distance (eV/atom) Num_atom
43 0.000000 (0, 2, 5)
42 0.000000 (0, 5, 5)
48 0.000000 (0, 1, 5)
46 0.000009 (0, 1, 5)
28 0.000011 (0, 1, 5)
41 0.000360 (0, 4, 6)
47 0.001838 (0, 1, 5)
36 0.001992 (1, 1, 6)
21 0.002544 (0, 1, 7)
23 0.002551 (0, 1, 7)
24 0.002795 (0, 4, 7)
例えば第3世代終了時点の凸包プロットの画像データは./convex_hull/conv_hull_gen_3.svgに出力される.
デフォルトはsvg形式であるが,入力ファイルでfig_formatを変更していればpdfやpngで出力される.

EA-vcでは2元系および3元系の探索シミュレーションの場合,凸包のグラフは各世代の計算終了時に自動プロットされる. 自分でもっと編集したい場合はJupyterでカスタマイズできる. 4元系の時はJupyterを用いたplotly(pymatgenに必要なのでインストールされているはず)の可視化が利用可能. ここではいくつか例を示しておく.

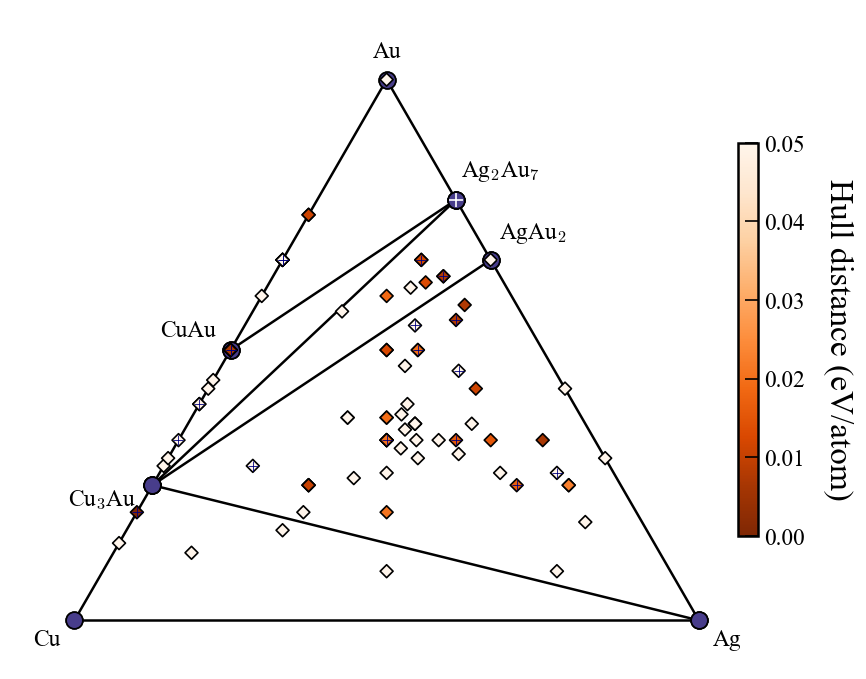
上図は3世代まで探索した時の例で,赤文字は説明のために追加した. 凸包プロットに関する入力ファイルの設定は以下が関係する.括弧内はデフォルト値.
show_max:y軸上限(0.2)label_stable:安定相の組成を表示するかどうか(True)vmax:右のカラーバーの最大値(0.2)bottom_margin:最小値とy軸下限の間のマージン(0.02).fig_format:図のファイルフォーマット.svg, png, pdfに対応.(svg)マーカー上の十字の印は最新世代の探索結果を示している.

上図は3世代まで探索した時の例で,赤文字は説明のために追加した. 凸包プロットに関する入力ファイルの設定は以下が関係する.括弧内はデフォルト値.
show_max:hull distanceがshow_max以下のものだけをプロット(0.2)label_stable:安定相の組成を表示するかどうか(True)vmax:右のカラーバーの最大値(0.2)bottom_margin:3元系では無関係fig_format:図のファイルフォーマット.svg, png, pdfに対応.(svg)マーカーの十字の印は最新世代の探索結果を示している.
ここでは,CrySPYのデータをローカルPCで解析・可視化することを前提としている.
CrySPYをスーパーコンピュータやワークステーションで使用している場合は,データをローカルPCにダウンロードすること.
work や backup ディレクトリは,ファイルサイズが非常に大きくなる可能性があるため,不要であれば削除してよい.
先ほどダウンロードした結果の中にある data/ ディレクトリに移動する.
その後,CrySPY utilityがローカルにダウンロード してある場合は cryspy_analyzer_EA-vc.ipynb をコピーする.
またはGitHubから直接ダウンロードしてくる(CrySPY_utility/notebook/).
Jupyter notebookファイルには,CrySPYのコードと同じ関数が書かれており,自由に凸包のプロットをカスタマイズできる. 適宜順番に実行していき,下記のどちらかを選ぶと自動プロットと同じものが得られる
途中にある
では,2元系,3元系および4元系において,Plotlyを用いたインタラクティブプロットができる. プロット例はCrySPY > チュートリアル > インタラクティブモード(Jupyter Notebook) #Interactive plot using Plotlyを参考にすること.
BO
May 15th, 2023
CrySPYの基本的な使い方に関してははじめにTutorial > Random Search (RS)を見ること.
ここではCrySPY 1.1.0以上を想定している.
ここで利用しているファイルはCrySPY_utility/examples/qe_Si16_LAQAからダウンロードできる. このチュートリアルでは,50個だけ初期構造を生成しているが,本来LAQAでは,もっと多くの構造を生成しておいてそこから良い候補を選択することでシミュレーションを進める.
cryspy.inの例.
[basic]
algo = LAQA
calc_code = QE
tot_struc = 50
nstage = 1
njob = 10
jobcmd = qsub
jobfile = job_cryspy
[structure]
atype = Si
nat = 16
mindist_1 = 1.5
[QE]
qe_infile = pwscf.in
qe_outfile = pwscf.out
kppvol = 80
[LAQA]
nselect_laqa = 4
[option]
nstageは1にする必要がある.nselect_laqaだけ新しく設定する必要がある. nselect_laqaは一回の選択で選ばれる候補の数.下記のようにwfやwsを指定すれば,LAQAのスコアにおける重みも変えられる.
省略した場合,デフォルトでは0.1と10.0がそれぞれ使われる.
スコアの詳細についてはSearch algorithms > LAQAを見ること.
[LAQA]
nselect_laqa = 4
wf = 0.1
ws = 10.0
&control
calculation = 'vc-relax'
pseudo_dir = '/usr/local/gbrv/all_pbe_UPF_v1.5/'
outdir='./outdir/'
nstep = 10
/
&system
ibrav = 0
nat = 16
ntyp = 1
ecutwfc = 40
ecutrho = 200
occupations = 'smearing'
degauss = 0.01
/
&electrons
/
&ions
/
&cell
/
ATOMIC_SPECIES
Si -1.0 si_pbe_v1.uspp.F.UPF
nstepで1回の選択で何ステップ構造最適化を進めるかをコントロールする.(VASPではNSW)#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
####$ -V -S /bin/zsh
#$ -N Si_CrySPY_ID
#$ -pe smp 20
####$ -q ibis1.q
####$ -q ibis2.q
mpirun -np $NSLOTS pw.x -nk 4 < pwscf.in > pwscf.out
if [ -e "CRASH" ]; then
sed -i -e '3 s/^.*$/skip/' stat_job
exit 1
fi
sed -i -e '3 s/^.*$/done/' stat_job
自動化スクリプトも用意してある.このページの最下部参照.
cryspyと打って1回目の実行.
cryspy &
この入力ファイルではまず50構造生成されるのでlog_cryspyを見て確認する.
2023/05/13 13:02:07
CrySPY 1.1.0
Start cryspy.py
Number of MPI processes: 1
Read input file, cryspy.in
Save input data in cryspy.stat
# --------- Generate initial structures
# ------ mindist
Si - Si 1.5
Structure ID 0 was generated. Space group: 165 --> 165 P-3c1
Structure ID 1 was generated. Space group: 66 --> 66 Cccm
Structure ID 2 was generated. Space group: 146 --> 146 R3
Structure ID 3 was generated. Space group: 82 --> 82 I-4
Structure ID 4 was generated. Space group: 162 --> 162 P-31m
...
...
...
Structure ID 47 was generated. Space group: 90 --> 90 P42_12
Structure ID 48 was generated. Space group: 214 --> 214 I4_132
Structure ID 49 was generated. Space group: 23 --> 23 I222
Elapsed time for structure generation: 0:00:10.929030
# ---------- Initialize LAQA
# ---------- Selection 0
selected_id: 50 IDs
LAQAでは,はじめに全ての初期構造の最適化ジョブを実行する.
完全に最適化を終わらせるわけではなく,ここではnstep = 10にしているので,10ステップだけ実行される.
cryspyコマンドを繰り返して,初期構造全てについて10ステップの最適化を完了させる.
必要であれば,njobの値を上げておけば一度に多くのジョブがサブミットされる.
初めの最適化が全て終わると, log_cryspyの最後にLAQA is readyと表示される.
2023/05/13 13:23:31
CrySPY 1.1.0
Restart cryspy.py
Number of MPI processes: 1
# ---------- job status
ID 41: Stage 1 Done!
LAQA is ready
この状態でcryspy を実行すると,最初の選択が始まる.
2023/05/13 13:23:33
CrySPY 1.1.0
Restart cryspy.py
Number of MPI processes: 1
# ---------- job status
Backup data
# ---------- Selection 1
selected_id: 37 8 10 48
nselect_laqaで設定された構造の数だけ選択される.
cryspyをもう一度実行するとそれらのジョブ(次の10ステップ)がサブミットされる.
cryspy &
2023/05/13 13:23:36
CrySPY 1.1.0
Restart cryspy.py
Number of MPI processes: 1
# ---------- job status
ID 37: submit job, Stage 1
ID 8: submit job, Stage 1
ID 10: submit job, Stage 1
ID 48: submit job, Stage 1
あとはこれを何度も繰り返し行うことでスコアに応じて選択された構造の最適化が10ステップずつ進行する. ある程度の構造の最適化が完全に完了するまで進めて,止めたいタイミングでストップする.
シミュレーションの途中でスコアの確認がしたければ次のファイルを見ると良い:
他にもLAQAに関数ファイルがいくつか出力される:
ここではCrySPYのデータをローカルPCで解析する.
スパコンやワークステーションで計算を行ったら,ローカルPCにデータをダウンロードしておく.
今後必要なければ,ファイルサイズが大きいworkとbackupディレクトリは削除しておいて良い.
pklデータはgzipしておくとファイルサイズを減らすことができる.
ダウンロードした結果のdata/ディレクトリに移動して,cryspy_analyzer_LAQA.ipynbをCrySPY utilityからコピーする.
このjupyter notebookを順番に実行していけば下記のようなグラフとgif画像が作成できる. この例では,アニメーションのために全ての構造の最適化を完全に完了させた. (全て最適化を完了させるとランダムサーチと計算量が変わらないのでLAQAの優位性はない)
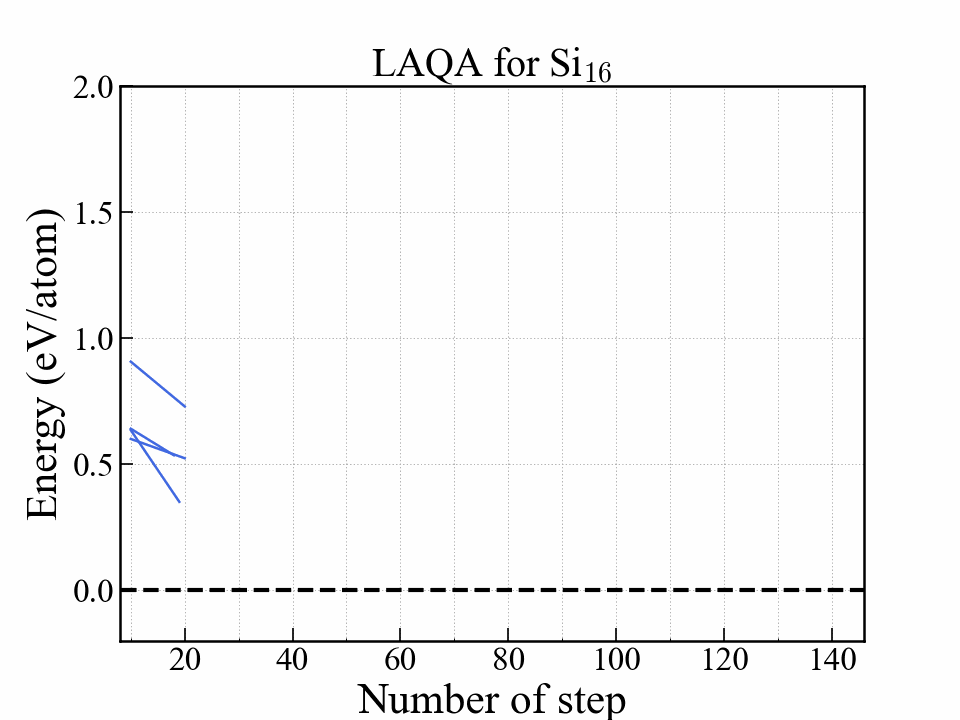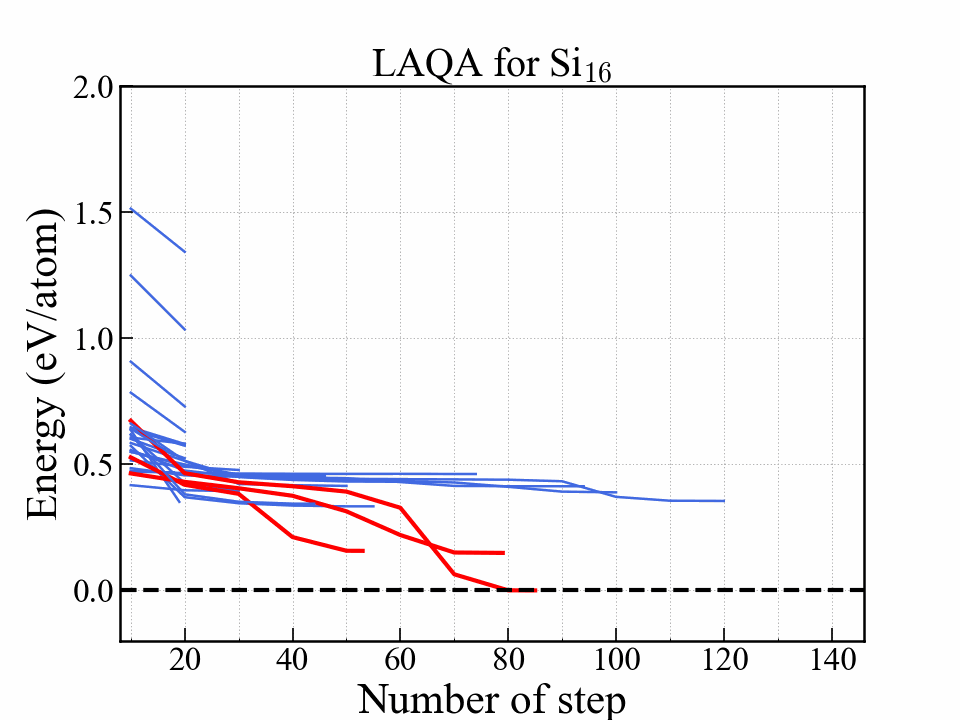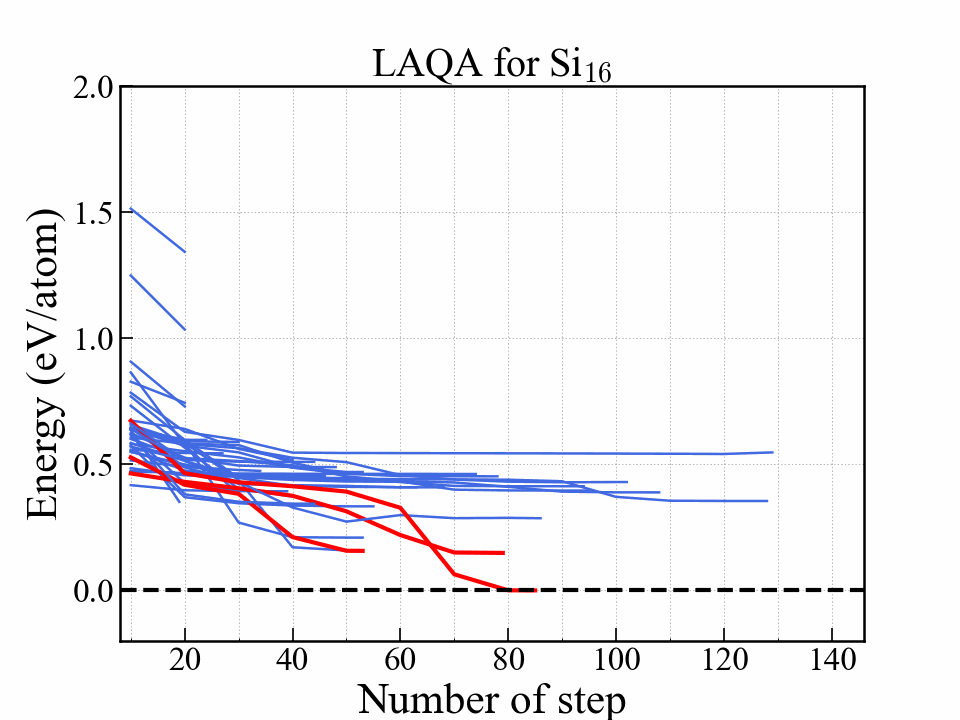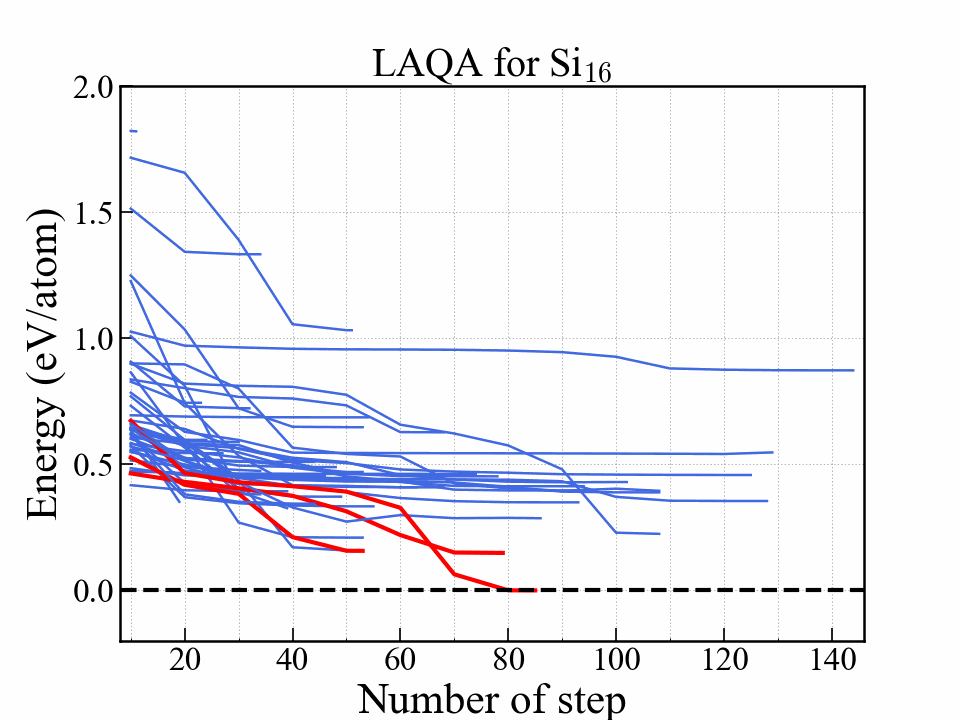
このグラフはエネルギーを最適化ステップの関数として示している. 赤い線は最終的にエネルギーが低かった3つの構造を表しており,中でも一番安定だった構造はダイアモンド構造に到達している. 安定になる構造はかなり早い段階で選択されて構造最適化が完了していることがわかる.
algo = LAQAでは[option]セクションの下記の二つは自動的にTrueになる.
原子に働く力とストレスのデータは1ステップごとに収集される. エネルギーと構造データは1ステップごとではなく,選択ごとに収集される. つまり,この場合は10ステップおきにエネルギーと構造データは保存される. もし1ステップごとのデータが欲しいのであれば,手動で下記の設定を追加すること.
[option]
energy_step_flag = True
struc_step_flag = True
何度も繰り返しcryspyを実行するのは面倒に感じたかもしれない. 下記のようなスクリプトを使えば自動化できる.
First, see Tutorial > Random Search (RS) for basic usage of CrySPY.
In this section, we give a tutorial on the molecular structure generation part only. Since version 0.9.0, CrySPY has been able to generate random molecular crystal structures using PyXtal.
You need to use a pre-defined molecular by PyXtal’s database (see, https://pyxtal.readthedocs.io/en/latest/Usage.html?highlight=benzene#pyxtal-molecule-pyxtal-molecule)) or create molecule files that define molecular structures.
PyXtal currently supports C60, H2O, CH4, NH3, benzene, naphthalene, anthracene, tetracene, pentacene, coumarin, resorcinol, benzamide, aspirin, ddt, lindane, glycine, glucose, and ROY.
Let us generate molecular crystal structures that consist of 2 benzenes.
Move to your working directory, and copy input example files by one of the following methods.
cp -r ~/CrySPY_root/CrySPY-0.9.0/example/QE_benzene_2_RS_mol .Take a look at cryspy.in.
$ cat cryspy.in
[basic]
algo = RS
calc_code = QE
tot_struc = 6
nstage = 2
njob = 2
jobcmd = qsub
jobfile = job_cryspy
[structure]
struc_mode = mol
atype = H C
nat = 12 12
mol_file = benzene
nmol = 2
[QE]
qe_infile = pwscf.in
qe_outfile = pwscf.out
kppvol = 40 60
[option]
In generating molecular crystal structures, you have to set struc_mode = mol in the [structure] section.
Molecule file(s) and the number of molecule(s) are specified as:
Run CrySPY and see the initial structures (./data/init_POSCARS).
Move to your working directory, and copy input example files for 2 formula units of Li3PS4.
cp -r ~/CrySPY_root/CrySPY-0.9.0/example/QE_Li3PS4_2fu_RS_mol .$ cd QE_Li3PS4_2fu_RS_mol
$ ls
Li.xyz PS4.xyz calc_in/ cryspy.in
Molecule files of Li and PS4 are included. Supported formats in PyXtal are .xyz, .gjf, .g03, .g09, .com, .inp, .out, and pymatgen’s JSON serialized molecules.
$ cat Li.xyz
1
New structure
Li 0.000 0.000 0.000
$ cat PS4.xyz
5
New structure
P 0.000000 0.000000 0.000000
S 1.200000 1.200000 -1.200000
S 1.200000 -1.200000 1.200000
S -1.200000 1.200000 1.200000
S -1.200000 -1.200000 -1.200000
Check cryspy.in.
$ cat cryspy.in
[basic]
algo = RS
calc_code = QE
tot_struc = 4
nstage = 2
njob = 1
jobcmd = qsub
jobfile = job_cryspy
[structure]
struc_mode = mol
atype = Li P S
nat = 6 2 8
mol_file = ./Li.xyz ./PS4.xyz
nmol = 6 2
[QE]
qe_infile = pwscf.in
qe_outfile = pwscf.out
kppvol = 40 60
[option]
A single atom (Li atom in this case) is treated as a molecule in the molecular crystal structure generation mode. In this example, a random molecular structure is composed of six Li molecules (atoms) and two PS4 molecules specified as:
In mol_file, set relative path of molecule files from cryspy.in.
Here the molecule files are placed in the same directory.
Run CrySPY and see the initial structures (./data/init_POSCARS).
Molecular crystal structure generation can be time consuming because PyXtal calculates the molecule directions according to a specified space group.
Sometimes molecular crystal structure generation gets stuck.
So we set a time limit on the single structure generation.
The time limit (timeout_mol) is set to 120 seconds by default.
If the limit is insufficient, you have to increase it as (see last line):
struc_mode = mol
atype = Li P S
nat = 6 2 8
mol_file = ./Li.xyz ./PS4.xyz
nmol = 6 2
timeout_mol = 300.0
You can control the volume of unit cells by changing the value(s) of scaling factor, vol_factor, in cryspy.in.
By default, vol_factor is set to 1.0.
It is also possible to specify a range of factors.
Set minimum and maximum values as follows:
struc_mode = mol
atype = Li P S
nat = 6 2 8
mol_file = ./Li.xyz ./PS4.xyz
nmol = 6 2
timeout_mol = 300.0
vol_factor = 0.8 1.5
2023/10/21 update
CrySPYの基本的な使い方に関してはチュートリアル > Random Search (RS)を見ること.
動作環境:
1.1.0 <= CrySPY <=1.2.2ではバグがあった.
MPIを使ったジョブをbashやzshで実行するとき(e.g., jobcmd = zsh, jobfile = job_cryspy),MPIのジョブが流れない.
qsubやsbatchでジョブスケジューラーを使う場合は問題ない。
このバグはバージョン1.2.3で修正.
mpi4pyのインストールがまだであればインストールする.
pip install mpi4py
cryspy.inはいつもと同じで変更する必要はない.ここでは下記の設定でMPIを使った構造生成を行う.
[basic]
algo = RS
calc_code = soiap
tot_struc = 100
nstage = 1
njob = 2
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Si
nat = 8
[soiap]
soiap_infile = soiap.in
soiap_outfile = soiap.out
soiap_cif = initial.cif
[option]
tot_struc,atype,nat以外の変数は構造生成に関係がないのでここでは無視して良い.
4並列で実行する場合,mpiexec -nを使う.-pオプションも必要.
mpiexec -n 4 cryspy -p
In 1.1.0 <= CrySPY <= 1.2.2では下記のコマンド (-pオプションは使用しない)
mpiexec -n 4 cryspy
ジョブスケジューラーなどにサブミットするときは自分でジョブファイルを作る.下記は一例.
#!/bin/sh
#$ -cwd
#$ -V -S /bin/bash
#$ -N n_nproc
#$ -pe smp 4
mpirun -np $NSLOTS ~/.local/bin/cryspy
実行スクリプトcryspyのpathなどは適宜編集すること.
CrySPYはシンプルに構造生成タスクをプロセス数で分割している.
構造が生成された順番でログが出力される.
2023/04/24 22:47:51
CrySPY 1.1.0
Start cryspy.py
Number of MPI processes: 4
Read input file, cryspy.in
Save input data in cryspy.stat
# --------- Generate initial structures
# ------ mindist
Si - Si 1.11
Structure ID 25 was generated. Space group: 138 --> 123 P4/mmm
Structure ID 75 was generated. Space group: 99 --> 99 P4mm
Structure ID 0 was generated. Space group: 127 --> 123 P4/mmm
Structure ID 1 was generated. Space group: 61 --> 61 Pbca
Structure ID 50 was generated. Space group: 38 --> 38 Amm2
Structure ID 51 was generated. Space group: 134 --> 123 P4/mmm
Structure ID 26 was generated. Space group: 111 --> 123 P4/mmm
Structure ID 2 was generated. Space group: 9 --> 9 Cc
Structure ID 3 was generated. Space group: 80 --> 80 I4_1
Structure ID 4 was generated. Space group: 107 --> 107 I4mm
Structure ID 5 was generated. Space group: 75 --> 75 P4
Structure ID 76 was generated. Space group: 108 --> 108 I4cm
Structure ID 77 was generated. Space group: 100 --> 100 P4bm
Structure ID 27 was generated. Space group: 207 --> 221 Pm-3m
しかし,init_POSCARSでは,構造生成が全て終わった後に出力しているのでID順になっている.
ID_0
1.0
2.9636956737951818 0.0000000000000002 0.0000000000000002
0.0000000000000000 2.9636956737951818 0.0000000000000002
0.0000000000000000 0.0000000000000000 6.2634106638053080
Si
8
direct
-0.1602734164607877 -0.1602734164607877 -0.0000000000000000 Si
0.1602734164607877 0.1602734164607877 0.5000000000000000 Si
0.6602734164607877 0.3397265835392123 0.7500000000000000 Si
0.3397265835392122 0.6602734164607877 0.2500000000000000 Si
0.4469739273741755 0.4469739273741755 -0.0000000000000000 Si
0.5530260726258245 0.5530260726258244 0.5000000000000000 Si
0.0530260726258245 0.9469739273741754 0.7500000000000000 Si
0.9469739273741754 0.0530260726258245 0.2500000000000000 Si
ID_1
1.0
7.2751506682509657 0.0000000000000004 0.0000000000000004
0.0000000000000000 7.2751506682509657 0.0000000000000004
0.0000000000000000 0.0000000000000000 5.1777634169924873
Si
8
direct
-0.3845341807505553 -0.3845341807505553 0.4999999999999999 Si
0.3845341807505553 0.3845341807505553 0.5000000000000000 Si
0.3845341807505553 -0.3845341807505553 0.0000000000000000 Si
-0.3845341807505553 0.3845341807505553 -0.0000000000000000 Si
0.0000000000000000 0.5000000000000000 0.2500000000000000 Si
0.5000000000000000 0.0000000000000000 0.7500000000000000 Si
0.0000000000000000 0.5000000000000000 0.7500000000000000 Si
0.5000000000000000 0.0000000000000000 0.2500000000000000 Si
ID_2
1.0
-4.3660398676292269 -4.3660398676292269 0.0000000000000000
-4.3660398676292269 -0.0000000000000003 -4.3660398676292269
0.0000000000000000 -4.3660398676292269 -4.3660398676292269
Si
8
direct
0.8700001548800920 0.8700001548800920 0.1299998451199080 Si
0.1299998451199080 0.1299998451199080 0.8700001548800920 Si
0.8700001548800920 0.1299998451199080 0.8700001548800920 Si
0.1299998451199080 0.8700001548800920 0.1299998451199080 Si
0.1299998451199080 0.8700001548800920 0.8700001548800920 Si
0.8700001548800920 0.1299998451199080 0.1299998451199080 Si
0.7500000000000000 0.7500000000000000 0.7500000000000000 Si
0.2500000000000000 0.2500000000000000 0.2500000000000000 Si
ランダム構造生成以外の部分はMPIを使っても並列化されていないので意味はない.
2025年3月6日
CrySPYをインストールすればASEは自動的にインストールされている. ワークステーションやローカルPCでJupyterを使えるようにしておく. このチュートリアルでは,構造最適化にPure Python EMT calculatorを用いる.このEMTポテンシャルの精度は悪く,デモ用のものなので注意.
exampleのノートブックには汎用機械学習ポテンシャルのCHGNetを使用するコードも書いてあるので,CHGNetを試したい場合は事前にpipでインストールしておく.
どこか適当なワーキングディレクトリに移動して,まずはexampleをコピーしてくる.下記のどちらからコピー してきても良い.
インタラクティブモードでも,cryspy.inを入力ファイルとして使用する.
インタラクティブモードではcalc_inディレクトリは使用しない.
input_examplesディレクトリの中に,いくつかの入力ファイル例が入っているので参考にしてほしい.
ここではEA-vcを用いた下記のcryspy.inを使用する.EA-vcについてはEA-vcのチュートリアルを見ること.
[basic]
algo = EA-vc
calc_code = ASE
nstage = 1
njob = 10
jobcmd = zsh
jobfile = job_cryspy
[structure]
atype = Cu Au
ll_nat = 0 0
ul_nat = 8 8
[ASE]
ase_python = ase_in.py
[EA]
n_pop = 20
n_crsov = 5
n_perm = 2
n_strain = 2
n_rand = 2
n_add = 3
n_elim = 3
n_subs = 3
target = random
n_elite = 2
n_fittest = 10
slct_func = TNM
t_size = 2
maxgen_ea = 5
end_point = 0.0 0.0
[option]
cryspy_interactive.ipynbを開いて,上から実行していく.
最初のセルはファイルやcryspy.inの中身を確認しているだけ.
!pwd
print()
!ls
print()
!cat cryspy.in
コメントアウトされているものは今回は無視して,CrySPYのインタラクティブモードで核となるライブラリをインポートするセルを実行する.
# ---------- import
from cryspy.interactive import action
このセルは通常の初回実行に対応している.cryspy.inを読み込んで初期構造が生成される.
# ---------- initial structure generation
action.initialize()
このセルでASEのcalculatorをセットする.ここではASEのEMTを用いる.
# ---------- EMT in ASE
from ase.calculators.emt import EMT
calculator = EMT()
# ---------- CHGNet
#from chgnet.model import CHGNetCalculator
#calculator = CHGNetCalculator()
このセルを実行すると,先ほど生成した初期構造の最適化が始まる. インタラクティブモードでは一つ一つ順番に構造最適化計算を行う.その際に進捗バーも表示される.
# ---------- structure optimization
action.restart(
njob=20, # njob=0: njob in cryspy.in will be used
calculator=calculator,
optimizer='BFGS', # 'FIRE', 'BFGS' or 'LBFGS'
symmetry=True, # default: True
fmax=0.01, # default: 0.01 eV/Å
steps=2000, # default: 2000
)
cryspy.inに書いてある値が使用される.FIRE, BFGS, LBFGSから選択.文字列で指定する.njobの値を小さくしている場合は,何度かこのセルを実行して,全ての初期構造の最適化を終了させる.
EA-vcを用いている場合,すべて終わると以下のように表示される.
EA is ready
もう一度このセルを実行すると世代交代が行われるので,次世代の構造生成が終了したら,あとは同様にこのセルの実行を繰り返す.
このセルを実行すると,cryspy_rslt_energy_ascなどのファイルを表示できる.
# ---------- show results
#!cat ./data/cryspy_rslt # Order of structure optimization completion
!cat ./data/cryspy_rslt_energy_asc # show energy ascending order
#!sed -n 2,4p ./data/cryspy_rslt # show i--jth lines
#!tail -n 5 ./data/cryspy_rslt # show last 5 lines
初期構造や最適化済みの構造をインタラクティブに可視化できる.
from ase.visualize import view
atoms = action.get_atoms('opt', cid=0) # 'init' or 'opt'
view(atoms, viewer='ngl') # viewer = 'ngl', 'ase', or 'x3d'
action.get_atoms('opt', cid=0)のところでoptをinitに変えると,初期構造が確認できる.
cidで構造IDを指定する.
ASEの機能を使用しているのでviewerにはngl, ase, x3dが利用できる.
nglを使うにはnglviewのインストールが必要なのでpipでインストールしておく.
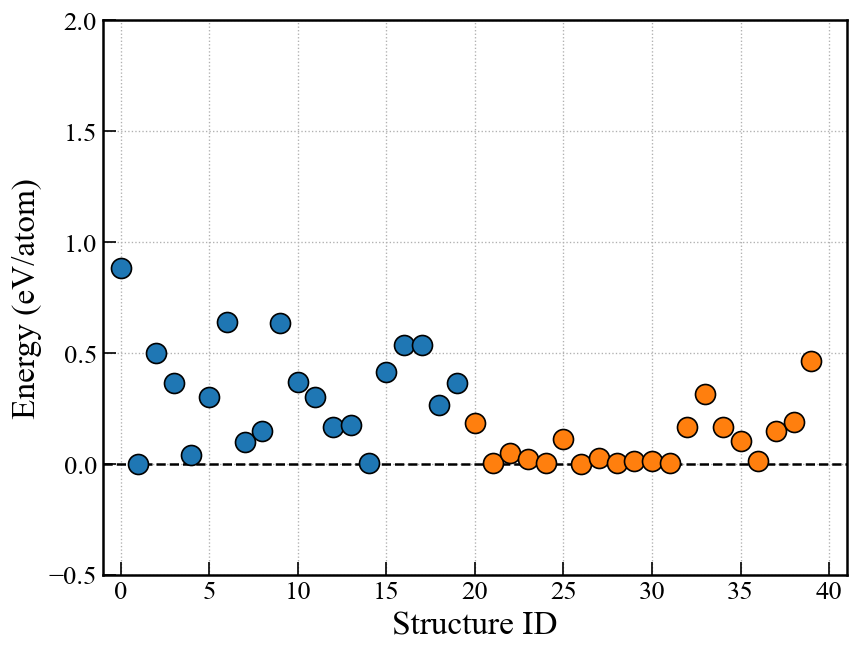
ランダムサーチ(RS)や進化的アルゴリズム(EA)の場合,下記画像のようなエネルギーグラフを表示できる. EA-vcの場合は原子数が異なるので単純にエネルギーを比較できないので,後述のconvex hullプロットを使う.
fig, ax = action.plot_E(
title=None,
ymax=2.0,
ymin=-0.5,
markersize=12,
marker_edge_width=1.0,
marker_edge_color='black',
alpha=1.0,
)
EA-vcの場合はPlotlyを用いたconvex hullのインタラクティブプロットが利用できる. CrySPYをインストールすればPlotlyは自動的にインストールされている. このconvex hullプロットはpymatgenの機能を用いている.
action.interactive_plot_convex_hull(cgen=None, show_unstable=0.2, ternary_style='2d')
2元系ではなく,3元系や4元系で計算しているときは下記のようなインタラクティブプロットが得られる. 左から順に,3元系(ternary_style = ‘2d’),3元系(ternary_style = ‘3d’),4元系(ternary_style = ‘3d’)のプロット
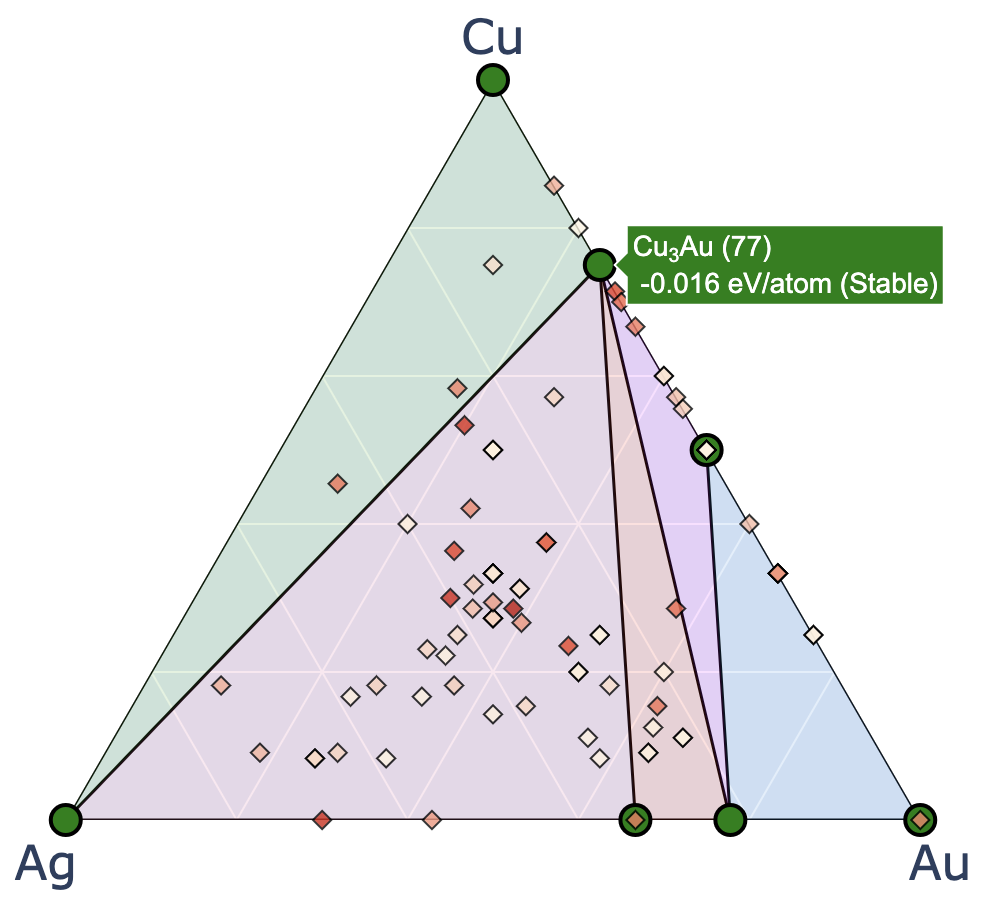
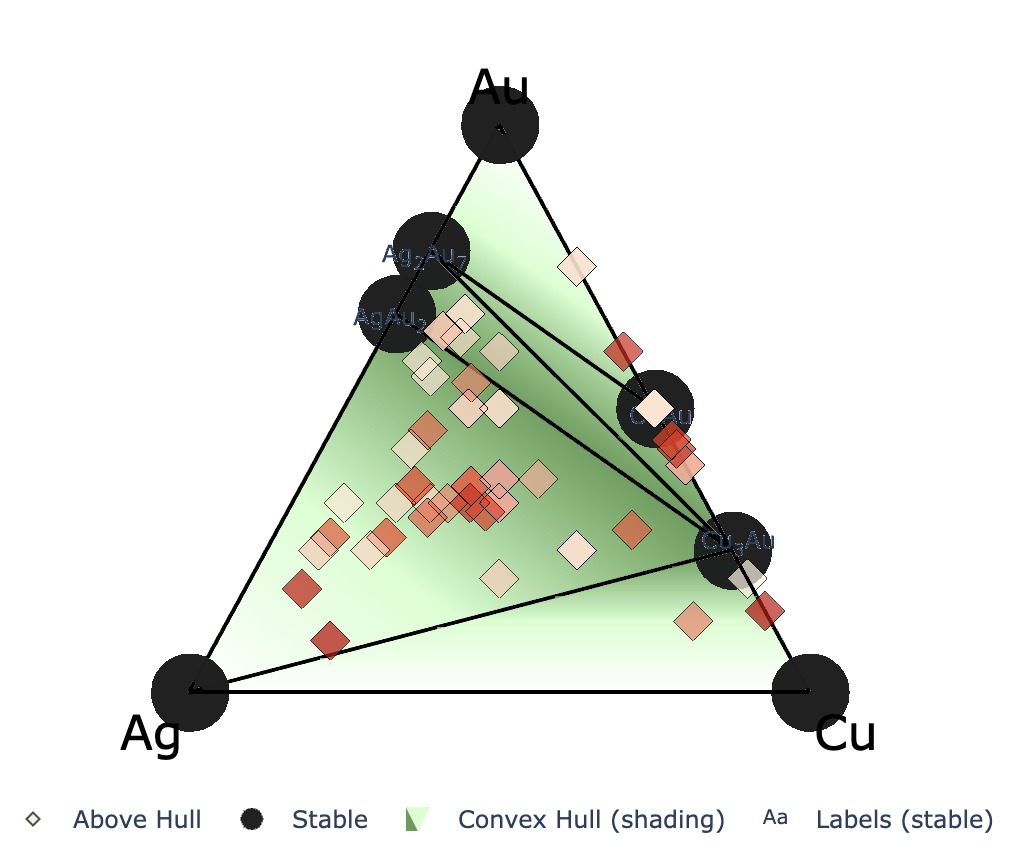
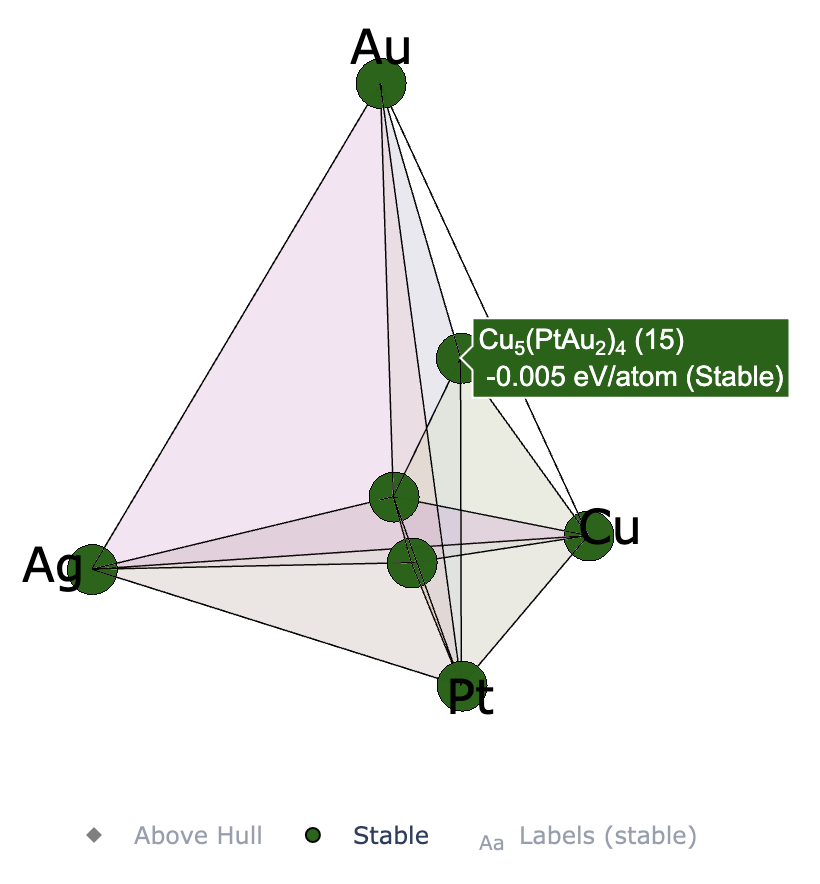
このセルを実行すると,matplotlibを用いて2元系のconvex hullをプロットできる.
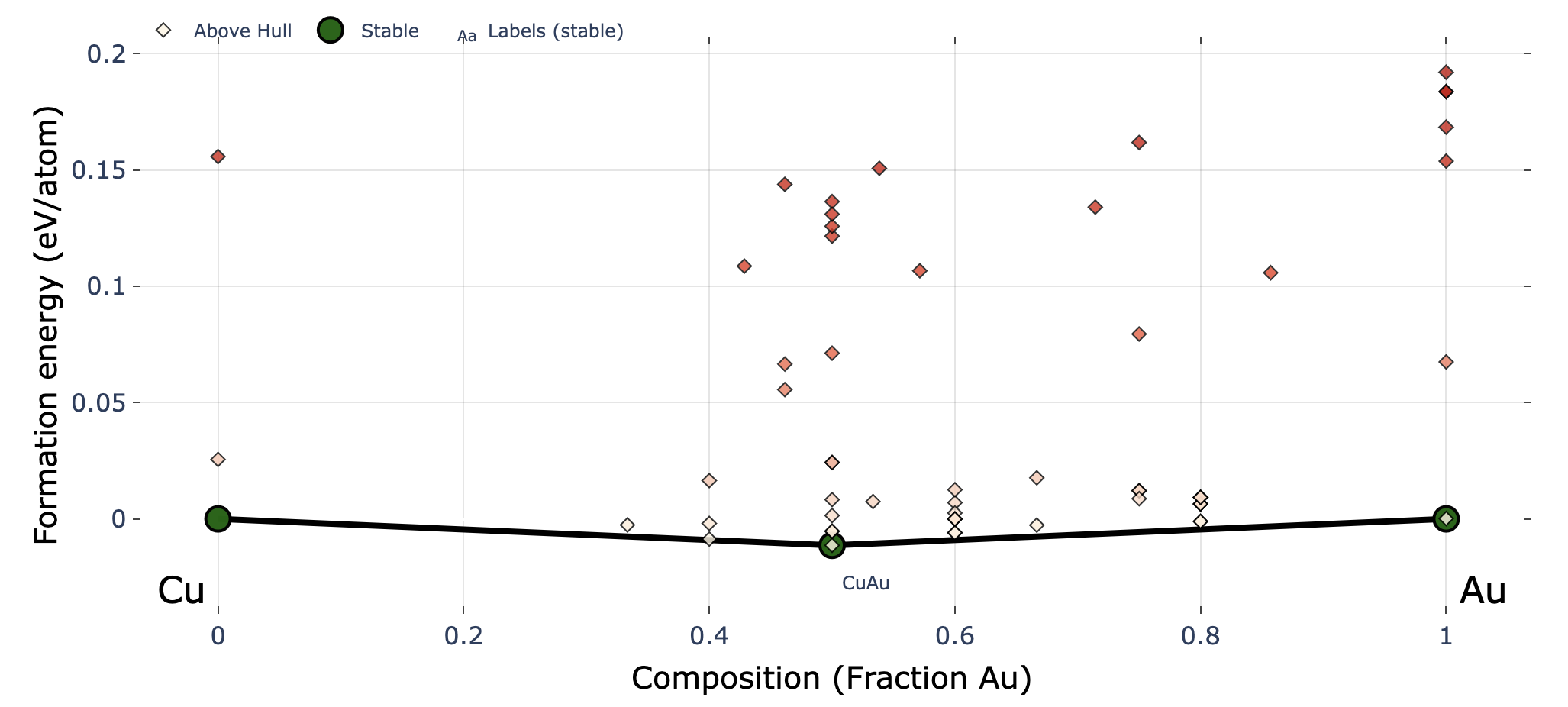
fig, ax = action.plot_convex_hull_binary(
cgen=None,
show_max=0.2,
label_stable=True,
vmax=0.2,
bottom_margin=0.02,
)
fig # to show plot in jupyter
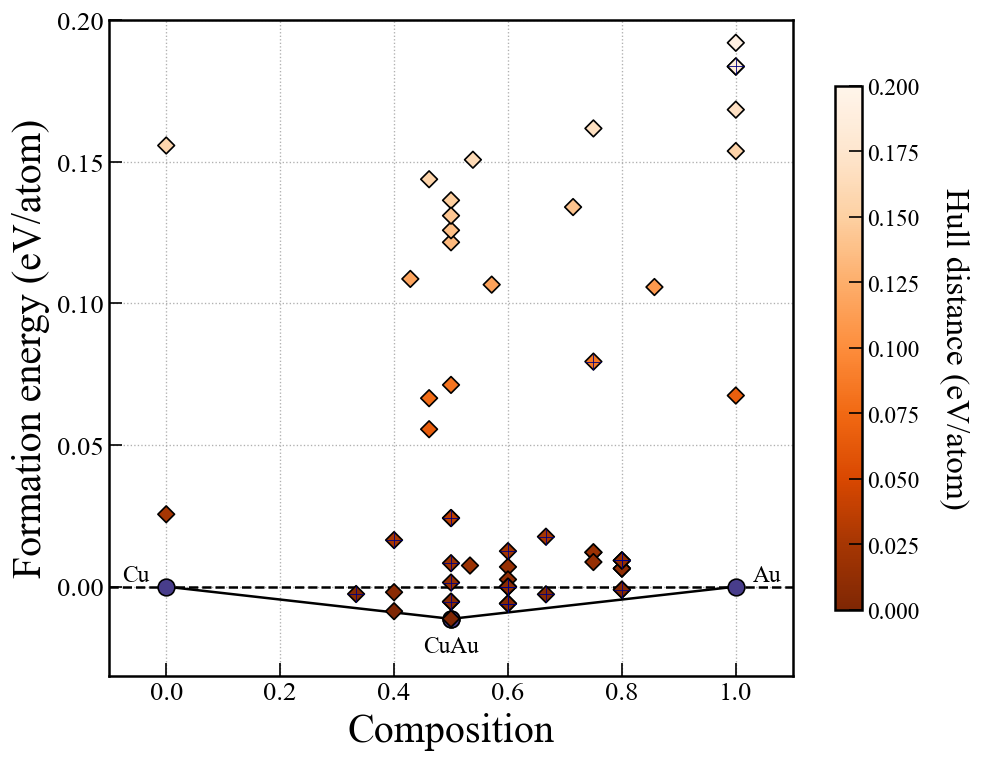
もし3元系の探索をしている場合は,このセルを実行することでmatplotlibを使ったconvex hullのプロットができる.
fig, ax = action.plot_convex_hull_ternary(
cgen=None,
show_max=0.2,
label_stable=True,
vmax=0.2,
)
fig # to show plot in jupyter
例えば以下のようなプロットが得られる.